

Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases



{kind=link}
{kind=link}
Abstract
1. Introduction
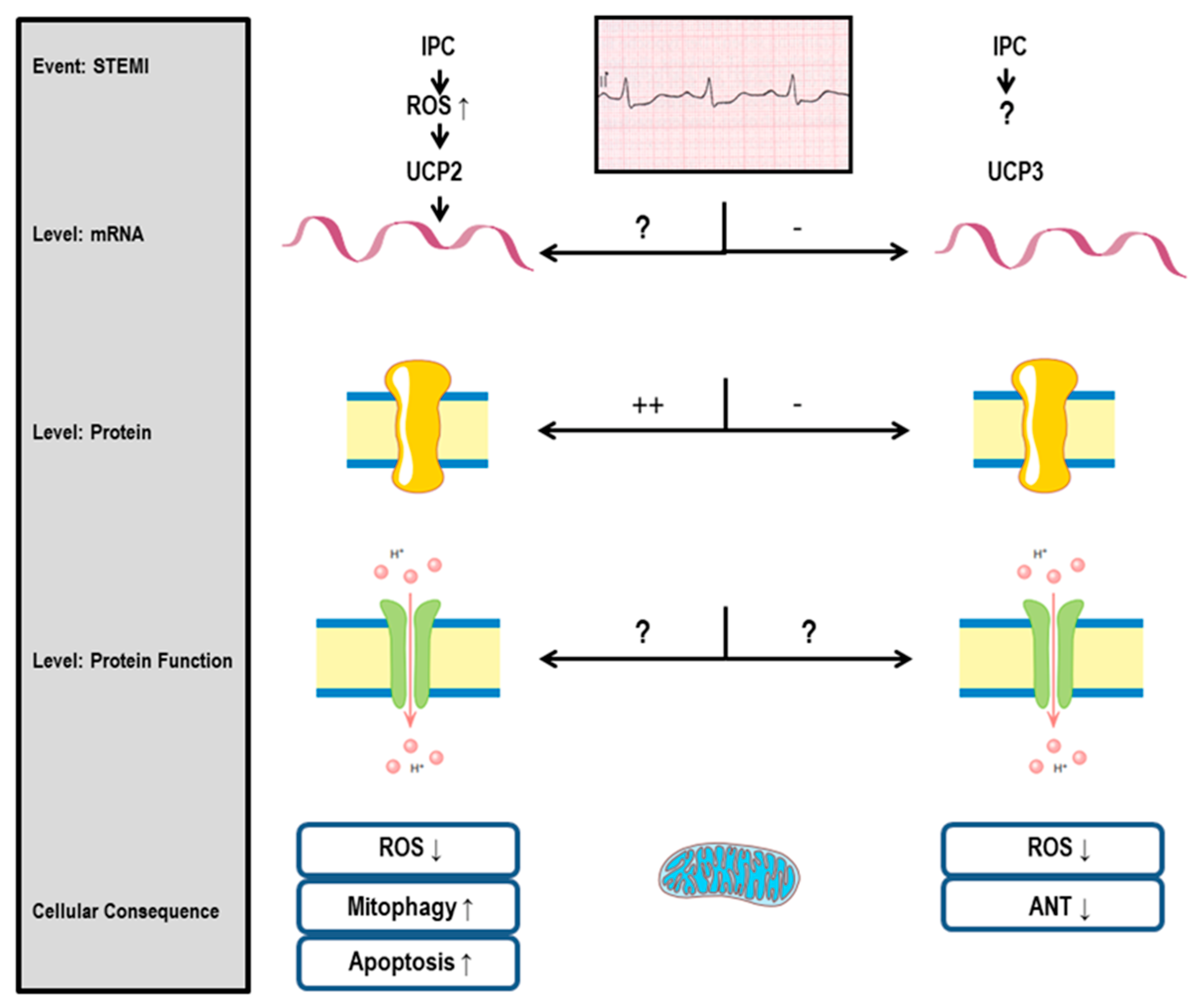
2. Uncoupling proteins (UCP)
2.1. Mitochondrial Reactive Oxygen Species and Cardiac Ischemia/Reperfusion Injury
2.2. Metabolism and Ischemia/Reperfusion Injury
3. Uncoupling Proteins
3.1. UCP2 and Cardiac Protection
3.2. UCP3 and Cardiac Protection
3.3. UCP2 and I/R Injury: General Aspects
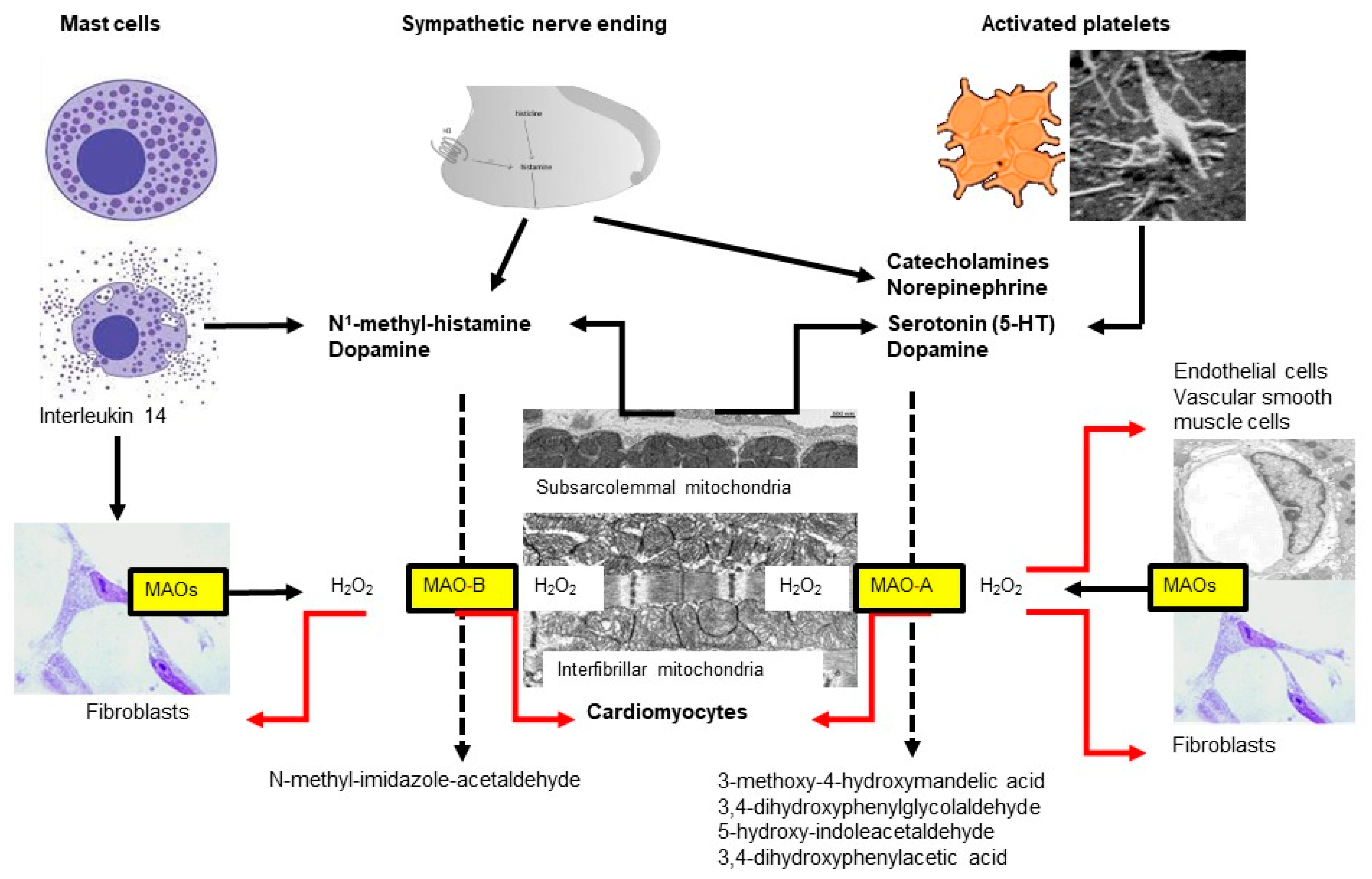
4. Monoamine Oxidases (MAO)
4.1. Monoamine Oxidase Isoforms
4.2. MAO Substrates
4.3. MAO Expression
4.4. Monoamine Oxidases and Hypertrophy
4.5. Pulmonary Hypertension
4.6. Monoamine Oxidases and Ischemia/Reperfusion (I/R) Injury (for Review Also See [134,169])
4.7. Monoamine Oxidases and Left Ventricular Remodeling/Heart Failure
5. Conclusions
Funding
Conflicts of Interest
References
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Santos, C.X.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell Cardiol. 2014, 73, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.T.; Rybin, V.; Marin-Garcia, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015, 20, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Efentakis, P.; Frenis, K.; Daiber, A.; Schulz, R. Thiol-based redox-active proteins as cardioprotective therapeutic agents in cardiovascular diseases. Basic Res. Cardiol. 2021, 116, 44. [Google Scholar] [CrossRef]
- Boengler, K.; Leybaert, L.; Ruiz-Meana, M.; Schulz, R. Connexin 43 in Mitochondria: What Do We Really Know About Its Function? Front. Physiol. 2022, 13, 928934. [Google Scholar] [CrossRef] [PubMed]
- Comita, S.; Femmino, S.; Thairi, C.; Alloatti, G.; Boengler, K.; Pagliaro, P.; Penna, C. Regulation of STAT3 and its role in cardioprotection by conditioning: Focus on non-genomic roles targeting mitochondrial function. Basic Res. Cardiol. 2021, 116, 56. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Bornbaum, J.; Schluter, K.D.; Schulz, R. P66shc and its role in ischemic cardiovascular diseases. Basic Res. Cardiol. 2019, 114, 29. [Google Scholar] [CrossRef]
- Schluter, K.D.; Kutsche, H.S.; Hirschhauser, C.; Schreckenberg, R.; Schulz, R. Review on Chamber-Specific Differences in Right and Left Heart Reactive Oxygen Species Handling. Front. Physiol. 2018, 9, 1799. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Piper, H.M.; Garcia-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord EN, J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Burger, N.; Kula-Alwar, D.; Aksentijevic, D.; Bridges, H.R.; Prag, H.A.; Grba, D.N.; Viscomi, C.; James, A.M.; Mottahedin, A.; et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 2021, 12, 707. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miro-Casas, E.; Alburquerque-Bejar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodriguez-Sinovas, A.; Garcia-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Prag, H.A.; Aksentijevic, D.; Dannhorn, A.; Giles, A.V.; Mulvey, J.F.; Sauchanka, O.; Du, L.; Bates, G.; Reinhold, J.; Kula-Alwar, D.; et al. Ischemia-Selective Cardioprotection by Malonate for Ischemia/Reperfusion Injury. Circ. Res. 2022, 131, 528–541. [Google Scholar] [CrossRef]
- Schulz, R.; Heusch, G. Targeted Mito- and Cardioprotection by Malonate. Circ. Res. 2022, 131, 542–544. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miro-Casas, E.; Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Garcia-Dorado, D. Selective Inhibition of Succinate Dehydrogenase in Reperfused Myocardium with Intracoronary Malonate Reduces Infarct Size. Sci. Rep. 2018, 8, 2442. [Google Scholar] [CrossRef]
- Consegal, M.; Nunez, N.; Barba, I.; Benito, B.; Ruiz-Meana, M.; Inserte, J.; Ferreira-Gonzalez, I.; Rodriguez-Sinovas, A. Citric Acid Cycle Metabolites Predict Infarct Size in Pigs Submitted to Transient Coronary Artery Occlusion and Treated with Succinate Dehydrogenase Inhibitors or Remote Ischemic Perconditioning. Int. J. Mol. Sci. 2021, 22, 4151. [Google Scholar] [CrossRef]
- Boengler, K.; Ungefug, E.; Heusch, G.; Schulz, R. The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr. Pharm. Des. 2013, 19, 6890–6895. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.; Szczepanek, K.; Bowler, E.; Hu, Y.; Larner, A.; Lesnefsky, E.J.; Chen, Q. Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: Role of complex I inhibition vs. depolarization of inner mitochondrial membrane. Biochim. Biophys. Acta 2013, 1830, 4537–4542. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, D.J.; Action, E.-C.C. Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef]
- Ong, S.B.; Kwek, X.Y.; Katwadi, K.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Ismail, N.I.; Lin, Y.H.; Yap, E.P.; Lim, S.Y.; Ja, K.; et al. Targeting Mitochondrial Fission Using Mdivi-1 in A Clinically Relevant Large Animal Model of Acute Myocardial Infarction: A Pilot Study. Int. J. Mol. Sci. 2019, 20, 3972. [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Mekki, M.; Corbiere, C.; Karoui, A.; Noel, R.; Vendeville, C.; Vaugeois, J.M.; Monteil, C. Subsarcolemmal and interfibrillar mitochondria display distinct superoxide production profiles. Free Radic. Res. 2015, 49, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. 1997, 273 Pt 2, H1544–H1554. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Nunez, E.; Miro-Casas, E.; Martinez-Acedo, P.; Barba, I.; Rodriguez-Sinovas, A.; Inserte, J.; Fernandez-Sanz, C.; Hernando, V.; Vazquez, J.; et al. Ischemic preconditioning protects cardiomyocyte mitochondria through mechanisms independent of cytosol. J. Mol. Cell. Cardiol. 2014, 68, 79–88. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef]
- Sodi-Pallares, D.; Testelli, M.R.; Fishleder, B.L.; Bisteni, A.; Medrano, G.A.; Friedland, C.; De Micheli, A. Effects of an intravenous infusion of a potassium-glucose-insulin solution on the electrocardiographic signs of myocardial infarction. A preliminary clinical report. Am. J. Cardiol. 1962, 9, 166–181. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef]
- Lochner, A.; Pentz, A.; Williams, K.; Tromp, E.; Harper, I.S. Substrate effects on sarcolemmal permeability in the normoxic and hypoxic perfused rat heart. Basic Res. Cardiol. 1996, 91, 64–78. [Google Scholar] [CrossRef]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Lochner, A.; Genade, S.; Genis, A.; Marais, E.; Salie, R. Long-chain free fatty acids inhibit ischaemic preconditioning of the isolated rat heart. Mol. Cell. Biochem. 2020, 473, 111–132. [Google Scholar] [CrossRef]
- Oeing, C.U.; Jun, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Chen, A.; Grajeda, M.I.; Tahir, U.A.; Gerszten, R.E.; Paolocci, N.; Ranek, M.J.; et al. MTORC1-Regulated Metabolism Controlled by TSC2 Limits Cardiac Reperfusion Injury. Circ. Res. 2021, 128, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Rey, B.; Sibille, B.; Romestaing, C.; Belouze, M.; Letexier, D.; Servais, S.; Barre, H.; Duchamp, C.; Voituron, Y. Reptilian uncoupling protein: Functionality and expression in sub-zero temperatures. J. Exp. Biol. 2008, 211 Pt 9, 1456–1462. [Google Scholar] [CrossRef]
- Voituron, Y.; Storey, J.M.; Grenot, C.; Storey, K.B. Freezing survival, body ice content and blood composition of the freeze-tolerant European common lizard, Lacerta vivipara. J. Comp. Physiol. B 2002, 172, 71–76. [Google Scholar]
- Kutsche, H.S.; Schreckenberg, R.; Schluter, K.D. Uncoupling Proteins in Striated Muscle Tissue: Known Facts and Open Questions. Antioxid. Redox. Signal. 2022, 37, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schluter, K.D. Specific Mechanisms Underlying Right Heart Failure: The Missing Upregulation of Superoxide Dismutase-2 and Its Decisive Role in Antioxidative Defense. Antioxid. Redox. Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, A.; Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Pak, O.; Kojonazarov, B.; Sydykov, A.; Hirschhauser, C.; Wolf, A.; Haag, D.; et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc. Res. 2019, 115, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Hirschhauser, C.; Rohrbach, S.; Li, L.; Niemann, B.; Schulz, R.; Schluter, K.D. Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2. Cells 2020, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, J.A.; Ziemba, E.A.; Colbert, R.; Anderson, L.B.; Sluiter, W.; Duncker, D.J.; Butterick, T.A.; Sikora, J.; Ward, H.B.; Kelly, R.F.; et al. Altered expression of mitochondrial electron transport chain proteins and improved myocardial energetic state during late ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1974–H1982. [Google Scholar] [CrossRef] [PubMed]
- McFalls, E.O.; Sluiter, W.; Schoonderwoerd, K.; Manintveld, O.C.; Lamers, J.M.; Bezstarosti, K.; van Beusekom, H.M.; Sikora, J.; Ward, H.B.; Merkus, D.; et al. Mitochondrial adaptations within chronically ischemic swine myocardium. J. Mol. Cell. Cardiol. 2006, 41, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Safari, F.; Bayat, G.; Shekarforoush, S.; Hekmatimoghaddam, S.; Anvari, Z.; Moghadam, M.F.; Hajizadeh, S. Expressional profile of cardiac uncoupling protein-2 following myocardial ischemia reperfusion in losartan- and ramiprilat-treated rats. J. Renin. Angiotensin. Aldosterone Syst. 2014, 15, 209–217. [Google Scholar] [CrossRef]
- Deng, M.; Wang, D.; He, S.; Xu, R.; Xie, Y. SIRT1 confers protection against ischemia/reperfusion injury in cardiomyocytes via regulation of uncoupling protein 2 expression. Mol. Med. Rep. 2017, 16, 7098–7104. [Google Scholar] [CrossRef]
- Chen, G.G.; Yan, J.B.; Wang, X.M.; Zheng, M.Z.; Jiang, J.P.; Zhou, X.M.; Cai, B.; Shen, Y.L. Mechanism of uncoupling protein 2-mediated myocardial injury in hypothermic preserved rat hearts. Mol. Med. Rep. 2016, 14, 1857–1864. [Google Scholar] [CrossRef]
- McLeod, C.J.; Aziz, A.; Hoyt, R.F., Jr.; McCoy, J.P., Jr.; Sack, M.N. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J. Biol. Chem. 2005, 280, 33470–33476. [Google Scholar] [CrossRef]
- Wu, H.; Ye, M.; Liu, D.; Yang, J.; Ding, J.W.; Zhang, J.; Wang, X.A.; Dong, W.S.; Fan, Z.X.; Yang, J. UCP2 protect the heart from myocardial ischemia/reperfusion injury via induction of mitochondrial autophagy. J. Cell. Biochem. 2019, 120, 15455–15466. [Google Scholar] [CrossRef]
- Cheng, J.; Nanayakkara, G.; Shao, Y.; Cueto, R.; Wang, L.; Yang, W.Y.; Tian, Y.; Wang, H.; Yang, X. Mitochondrial Proton Leak Plays a Critical Role in Pathogenesis of Cardiovascular Diseases. Adv. Exp. Med. Biol. 2017, 982, 359–370. [Google Scholar]
- Schreckenberg, R.; Klein, J.; Kutsche, H.S.; Schulz, R.; Gomori, K.; Bencsik, P.; Benczik, B.; Agg, B.; Saghy, E.; Ferdinandy, P.; et al. Ischaemic post-conditioning in rats: Responder and non-responder differ in transcriptome of mitochondrial proteins. J. Cell. Mol. Med. 2020, 24, 5528–5541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Cui, Z.; Zheng, Y.; Li, Q.; Xu, C.; Sheng, X.; Tao, M.; Xu, H. Fenofibrate protects against acute myocardial I/R injury in rat by suppressing mitochondrial apoptosis as decreasing cleaved caspase-9 activation. Cancer Biomark. 2017, 19, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, A.; Brauer, A.U.; Smorodchenko, A.; Goyn, J.; Hilse, K.E.; Shabalina, I.G.; Infante-Duarte, C.; Pohl, E.E. Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS ONE 2012, 7, e41406. [Google Scholar] [CrossRef]
- Modriansky, M.; Gabrielova, E. Uncouple my heart: The benefits of inefficiency. J. Bioenerg. Biomembr. 2009, 41, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, S.; Jiang, L.; Hou, J.; Yang, Z. Curcumin Improves Cardiopulmonary Resuscitation Outcomes by Modulating Mitochondrial Metabolism and Apoptosis in a Rat Model of Cardiac Arrest. Front. Cardiovasc. Med. 2022, 9, 908755. [Google Scholar] [CrossRef]
- Larbig, R.; Reda, S.; Paar, V.; Trost, A.; Leitner, J.; Weichselbaumer, S.; Motloch, K.A.; Wernly, B.; Arrer, A.; Strauss, B.; et al. Through modulation of cardiac Ca(2+) handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca(2+)-mediated arrhythmias. Exp. Physiol. 2017, 102, 650–662. [Google Scholar] [CrossRef]
- Gibb, A.A.; Epstein, P.N.; Uchida, S.; Zheng, Y.; McNally, L.A.; Obal, D.; Katragadda, K.; Trainor, P.; Conklin, D.J.; Brittian, K.R.; et al. Exercise-Induced Changes in Glucose Metabolism Promote Physiological Cardiac Growth. Circulation 2017, 136, 2144–2157. [Google Scholar] [CrossRef]
- Dyck, S.; Kataria, H.; Alizadeh, A.; Santhosh, K.T.; Lang, B.; Silver, J.; Karimi-Abdolrezaee, S. Perturbing chondroitin sulfate proteoglycan signaling through LAR and PTPsigma receptors promotes a beneficial inflammatory response following spinal cord injury. J. Neuroinflamm. 2018, 15, 90. [Google Scholar] [CrossRef]
- Wang, Y.N.; Gao, L.; Wu, S.Y.; Qin, S. Low-Dose 4-Hydroxy-2-Nonenal (HNE) Reperfusion Therapy Displays Cardioprotective Effects in Mice After Myocardial Infarction That Are Abrogated by Genipin. Med. Sci. Monit. 2018, 24, 3702–3709. [Google Scholar] [CrossRef]
- Aslan, G.; Gul, H.F.; Tektemur, A.; Sahna, E. Ischemic postconditioning reduced myocardial ischemia-reperfusion injury: The roles of melatonin and uncoupling protein 3. Anatol. J. Cardiol. 2020, 23, 19–27. [Google Scholar]
- Banke, N.H.; Lewandowski, E.D. Impaired cytosolic NADH shuttling and elevated UCP3 contribute to inefficient citric acid cycle flux support of postischemic cardiac work in diabetic hearts. J. Mol. Cell. Cardiol. 2015, 79, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lou, P.H.; Lucchinetti, E.; Zhang, L.; Affolter, A.; Gandhi, M.; Hersberger, M.; Warren, B.E.; Lemieux, H.; Sobhi, H.F.; Clanachan, A.S.; et al. Loss of Intralipid(R)- but not sevoflurane-mediated cardioprotection in early type-2 diabetic hearts of fructose-fed rats: Importance of ROS signaling. PLoS ONE 2014, 9, e104971. [Google Scholar] [CrossRef]
- Harmancey, R.; Vasquez, H.G.; Guthrie, P.H.; Taegtmeyer, H. Decreased long-chain fatty acid oxidation impairs postischemic recovery of the insulin-resistant rat heart. FASEB J. 2013, 27, 3966–3978. [Google Scholar] [CrossRef]
- Harmancey, R.; Haight, D.L.; Watts, K.A.; Taegtmeyer, H. Chronic Hyperinsulinemia Causes Selective Insulin Resistance and Down-regulates Uncoupling Protein 3 (UCP3) through the Activation of Sterol Regulatory Element-binding Protein (SREBP)-1 Transcription Factor in the Mouse Heart. J. Biol. Chem. 2015, 290, 30947–30961. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.S.; Ashraf, S.; Lomax, T.M.; Wiseman, J.M.; Hall, M.E.; Gava, F.N.; Hall, J.E.; Hosler, J.P.; Harmancey, R. Uncoupling protein 3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res. Cardiol. 2018, 113, 47. [Google Scholar] [CrossRef]
- Inserte, J.; Aluja, D.; Barba, I.; Ruiz-Meana, M.; Miro, E.; Poncelas, M.; Vilardosa, U.; Castellano, J.; Garcia-Dorado, D. High-fat diet improves tolerance to myocardial ischemia by delaying normalization of intracellular PH at reperfusion. J. Mol. Cell. Cardiol. 2019, 133, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; Zou, L.; Qu, J.; Litovsky, S.; Umeda, P.; Zhou, L.; Chatham, J.; Marsh, S.A.; Dell’Italia, L.J.; et al. High-fat, low-carbohydrate diet promotes arrhythmic death and increases myocardial ischemia-reperfusion injury in rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H598–H608. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.K.; Soares, V.M.; Bernardo, A.F.; Neves, F.A.; Mattos, A.B.; Guedes, R.M.; Cortez, E.; Andrade, D.C.; Lacerda-Miranda, G.; Garcia-Souza, E.P.; et al. Overnourishment during lactation induces metabolic and haemodynamic heart impairment during adulthood. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bernardo, E.; Anedda, A.; Sanchez-Perez, P.; Acosta-Iborra, B.; Cadenas, S. 4-Hydroxynonenal induces Nrf2-mediated UCP3 upregulation in mouse cardiomyocytes. Free Radic. Biol. Med. 2015, 88 Pt B, 427–438. [Google Scholar] [CrossRef]
- Anedda, A.; Lopez-Bernardo, E.; Acosta-Iborra, B.; Saadeh Suleiman, M.; Landazuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Vuk, J.; Anders, M.E.; Rhee, S.W. Self-paced polling increases medical student engagement in recorded lectures and improves examination performance. Adv. Physiol. Educ. 2022, 46, 728–734. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Galatou, E.; Panagiotidis, G.D.; Ravingerova, T.; Lazou, A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. Int. J. Mol. Sci. 2021, 22, 6399. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yucel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, J.; Zheng, Y.; Wang, J.; Wang, Z.; Gu, S.; Tan, J.; Jing, Q.; Yang, H. Uncoupling protein 3 mediates H2O2 preconditioning-afforded cardioprotection through the inhibition of MPTP opening. Cardiovasc. Res. 2015, 105, 192–202. [Google Scholar] [CrossRef]
- Klumpe, I.; Savvatis, K.; Westermann, D.; Tschope, C.; Rauch, U.; Landmesser, U.; Schultheiss, H.P.; Dorner, A. Transgenic overexpression of adenine nucleotide translocase 1 protects ischemic hearts against oxidative stress. J. Mol. Med. (Berl.) 2016, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Abdallah, Y.; Shahzad, T.; Klumpe, I.; Piper, H.M.; Schultheiss, H.P.; Schluter, K.D.; Schulz, R.; Euler, G.; Dorner, A. Transgenic overexpression of the adenine nucleotide translocase 1 protects cardiomyocytes against TGFbeta1-induced apoptosis by stabilization of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 2012, 53, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Palmeri, M.; Horvath, T.L.; Russell, K.S.; Russell, R.R., 3rd. Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1192–H1200. [Google Scholar] [CrossRef] [PubMed]
- Pohl, E.E.; Rupprecht, A.; Macher, G.; Hilse, K.E. Important Trends in UCP3 Investigation. Front. Physiol. 2019, 10, 470. [Google Scholar] [CrossRef]
- Haines, B.A.; Mehta, S.L.; Pratt, S.M.; Warden, C.H.; Li, P.A. Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. J. Cereb. Blood Flow Metab. 2010, 30, 1825–1833. [Google Scholar] [CrossRef]
- He, M.; Ma, Y.; Wang, R.; Zhang, J.; Jing, L.; Li, P.A. Deletion of Mitochondrial Uncoupling Protein 2 Exacerbates Mitochondrial Damage in Mice Subjected to Cerebral Ischemia and Reperfusion Injury under both Normo- and Hyperglycemic Conditions. Int. J. Biol. Sci. 2020, 16, 2788–2802. [Google Scholar] [CrossRef]
- Zhang, T.; He, M.T.; Zhang, X.P.; Jing, L.; Zhang, J.Z. Uncoupling Protein 2 Deficiency Enhances NLRP3 Inflammasome Activation Following Hyperglycemia-Induced Exacerbation of Cerebral Ischemia and Reperfusion Damage In Vitro and In Vivo. Neurochem. Res. 2021, 46, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Maroto Cicuendez, I.; Fernandez-Diaz, E.; Garcia-Garcia, J.; Jordan, J.; Fernandez-Cadenas, I.; Montaner, J.; Serrano-Heras, G.; Segura, T. The UCP2-866G/A Polymorphism Could be Considered as a Genetic Marker of Different Functional Prognosis in Ischemic Stroke After Recanalization. Neuromol. Med. 2017, 19, 571–578. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Castello-Ruiz, M.; Burguete, M.C.; Aliena-Valero, A.; Salom, J.B.; Torregrosa, G.; Garcia-Segura, L.M. Insight into the molecular sex dimorphism of ischaemic stroke in rat cerebral cortex: Focus on neuroglobin, sex steroids and autophagy. Eur. J. Neurosci. 2020, 52, 2756–2770. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Z.; Zhou, W.; Lu, C.; Ji, T.; Yang, W.; Jin, Z.; Tian, Y.; Lei, W.; Wu, S.; et al. SIRT1/PGC-1alpha signaling activation by mangiferin attenuates cerebral hypoxia/reoxygenation injury in neuroblastoma cells. Eur. J. Pharmacol. 2021, 907, 174236. [Google Scholar] [CrossRef]
- Li, C.; Sun, H.; Arrick, D.M.; Mayhan, W.G. Chronic nicotine exposure exacerbates transient focal cerebral ischemia-induced brain injury. J. Appl. Physiol. (1985) 2016, 120, 328–333. [Google Scholar] [CrossRef]
- Yang, L.; Ma, Y.M.; Shen, X.L.; Fan, Y.C.; Zhang, J.Z.; Li, P.A.; Jing, L. The Involvement of Mitochondrial Biogenesis in Selenium Reduced Hyperglycemia-Aggravated Cerebral Ischemia Injury. Neurochem. Res. 2020, 45, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Wu, H.Y.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Lin, T.K.; Liou, C.W.; Chuang, Y.C. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem. Biophys. Res. Commun. 2006, 351, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiao, L.; Hou, Y.; He, Q.; Zhu, J.; Li, Y.; Wu, J.; Zhao, J.; Yu, S.; Zhao, Y. Sestrin2 Silencing Exacerbates Cerebral Ischemia/Reperfusion Injury by Decreasing Mitochondrial Biogenesis through the AMPK/PGC-1alpha Pathway in Rats. Sci. Rep. 2016, 6, 30272. [Google Scholar] [CrossRef]
- Zhao, B.; Sun, L.K.; Jiang, X.; Zhang, Y.; Kang, J.; Meng, H.; Li, H.; Su, J. Genipin protects against cerebral ischemia-reperfusion injury by regulating the UCP2-SIRT3 signaling pathway. Eur. J. Pharmacol. 2019, 845, 56–64. [Google Scholar] [CrossRef]
- Le Minh, K.; Berger, A.; Eipel, C.; Kuhla, A.; Minor, T.; Stegemann, J.; Vollmar, B. Uncoupling protein-2 deficient mice are not protected against warm ischemia/reperfusion injury of the liver. J. Surg. Res. 2011, 171, 742–748. [Google Scholar] [CrossRef]
- Ninomiya, M.; Shirabe, K.; Shimada, M.; Terashi, T.; Maehara, Y. Role of UCP2 expression after hepatic warm ischemia-reperfusion in the rat. Gut Liver 2011, 5, 486–492. [Google Scholar] [CrossRef]
- t Hart, N.A.; van der Plaats, A.; Faber, A.; Leuvenink, H.G.; Olinga, P.; Wiersema-Buist, J.; Verkerke, G.J.; Rakhorst, G.; Ploeg, R.J. Oxygenation during hypothermic rat liver preservation: An in vitro slice study to demonstrate beneficial or toxic oxygenation effects. Liver Transpl. 2005, 11, 1403–1411. [Google Scholar] [CrossRef]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Eipel, C.; Hubschmann, U.; Abshagen, K.; Wagner, K.F.; Menger, M.D.; Vollmar, B. Erythropoietin as additive of HTK preservation solution in cold ischemia/reperfusion injury of steatotic livers. J. Surg. Res. 2012, 173, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, J.; Dahmen, U. Computational Modeling of Oxidative Stress in Fatty Livers Elucidates the Underlying Mechanism of the Increased Susceptibility to Ischemia/Reperfusion Injury. Comput. Struct. Biotechnol. J. 2018, 16, 511–522. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Chavin, K.D.; Fiorini, R.N.; Shafizadeh, S.; Cheng, G.; Wan, C.; Evans, Z.; Rodwell, D.; Polito, C.; Haines, J.K.; Baillie, G.M.; et al. Fatty acid synthase blockade protects steatotic livers from warm ischemia reperfusion injury and transplantation. Am. J. Transplant. 2004, 4, 1440–1447. [Google Scholar] [CrossRef]
- Fiorini, R.N.; Donovan, J.L.; Rodwell, D.; Evans, Z.; Cheng, G.; May, H.D.; Milliken, C.E.; Markowitz, J.S.; Campbell, C.; Haines, J.K.; et al. Short-term administration of (-)-epigallocatechin gallate reduces hepatic steatosis and protects against warm hepatic ischemia/reperfusion injury in steatotic mice. Liver Transpl. 2005, 11, 298–308. [Google Scholar] [CrossRef]
- Nii, A.; Utsunomiya, T.; Shimada, M.; Ikegami, T.; Ishibashi, H.; Imura, S.; Morine, Y.; Ikemoto, T.; Sasaki, H.; Kawashima, A. Hydrolyzed whey peptide-based diet ameliorates hepatic ischemia-reperfusion injury in the rat nonalcoholic fatty liver. Surg. Today 2014, 44, 2354–2360. [Google Scholar] [CrossRef]
- Bardallo, R.G.; Da Silva, R.T.; Carbonell, T.; Palmeira, C.; Folch-Puy, E.; Rosello-Catafau, J.; Adam, R.; Panisello-Rosello, A. Liver Graft Hypothermic Static and Oxygenated Perfusion (HOPE) Strategies: A Mitochondrial Crossroads. Int. J. Mol. Sci. 2022, 23, 5742. [Google Scholar] [CrossRef]
- Liu, W.; Fan, Y.; Ding, H.; Han, D.; Yan, Y.; Wu, R.; Lv, Y.; Zheng, X. Normothermic machine perfusion attenuates hepatic ischaemia-reperfusion injury by inhibiting CIRP-mediated oxidative stress and mitochondrial fission. J. Cell. Mol. Med. 2021, 25, 11310–11321. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Wang, H.; Cheng, R.; Gou, S.; Liu, T. Effect of target-directed regulation of uncoupling protein-2 gene expression on ischemia-reperfusion injury of hepatocytes. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2008, 28, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Feng, Z.; Gao, W.; Duan, Y.; Fan, G.; Geng, X.; Wu, B.; Li, K.; Liu, K.; Peng, C. Aucubin Attenuates Liver Ischemia-Reperfusion Injury by Inhibiting the HMGB1/TLR-4/NF-kappaB Signaling Pathway, Oxidative Stress, and Apoptosis. Front. Pharmacol. 2020, 11, 544124. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Polito, C.C.; Haines, J.K.; Shafizadeh, S.F.; Fiorini, R.N.; Zhou, X.; Schmidt, M.G.; Chavin, K.D. Decrease of intracellular ATP content downregulated UCP2 expression in mouse hepatocytes. Biochem. Biophys. Res. Commun. 2003, 308, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Cai, T.; Ke, Q.; Yuan, Q.; Luo, J.; Mao, X.; Jiang, L.; Cao, H.; Wen, P.; Zen, K.; et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol. 2019, 247, 392–405. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Li, T.; Feng, Y.; Li, W.; Zhu, X.; Gu, R.; Zhou, L. Uncoupling protein 2 prevents ischaemia reperfusion injury through the regulation ROS/NF-kappaB signalling in mice. Mol. Membr. Biol. 2019, 35, 51–59. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, T.; Xu, J.; Jiang, L.; Wu, J.; Sun, Q.; Zen, K.; Yang, J. UCP2 attenuates apoptosis of tubular epithelial cells in renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2017, 313, F926–F937. [Google Scholar] [CrossRef]
- Chen, K.; Xu, Z.; Liu, Y.; Wang, Z.; Li, Y.; Xu, X.; Chen, C.; Xia, T.; Liao, Q.; Yao, Y.; et al. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Sci. Transl. Med. 2017, 9, eaao6298. [Google Scholar] [CrossRef]
- Ren, Y.; Qiu, M.; Zhang, J.; Bi, J.; Wang, M.; Hu, L.; Du, Z.; Li, T.; Zhang, L.; Wang, Y.; et al. Low Serum Irisin Concentration Is Associated with Poor Outcomes in Patients with Acute Pancreatitis, and Irisin Administration Protects Against Experimental Acute Pancreatitis. Antioxid. Redox. Signal. 2019, 31, 771–785. [Google Scholar] [CrossRef]
- Huang, L.; Belousova, T.; Chen, M.; DiMattia, G.; Liu, D.; Sheikh-Hamad, D. Overexpression of stanniocalcin-1 inhibits reactive oxygen species and renal ischemia/reperfusion injury in mice. Kidney Int. 2012, 82, 867–877. [Google Scholar] [CrossRef]
- Ke, Q.; Yuan, Q.; Qin, N.; Shi, C.; Luo, J.; Fang, Y.; Xu, L.; Sun, Q.; Zen, K.; Jiang, L.; et al. UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 2020, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.S.; Huang, L.; Belousova, T.; Lu, L.; Yang, Y.; Reddel, R.; Chang, A.; Ju, H.; DiMattia, G.; Tong, Q.; et al. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J. Am. Soc. Nephrol. 2015, 26, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kurella, M.; Beato, F.; Min, H.; Ingelfinger, J.R.; Stears, R.L.; Swinford, R.D.; Gullans, S.R.; Tang, S.S. Monitoring changes in gene expression in renal ischemia-reperfusion in the rat. Kidney Int. 2002, 61, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ji, J.; Zhou, X.; Li, R. Irisin Pretreatment Protects Kidneys against Acute Kidney Injury Induced by Ischemia/Reperfusion via Upregulating the Expression of Uncoupling Protein 2. Biomed. Res. Int. 2020, 2020, 6537371. [Google Scholar] [CrossRef]
- Basler, J. Retooling a Safety Simulation Into a Rapid Assessment Simulation for Senior Students. Nurse Educ. 2022, 47, 41. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.J.; Koon Yan, P.; Venetti, M.C. Developmental aspects of rat heart monoamine oxidase. Biochem. Pharmacol. 1979, 28, 2337–2343. [Google Scholar] [CrossRef]
- Monassier, L.; Laplante, M.A.; Ayadi, T.; Doly, S.; Maroteaux, L. Contribution of gene-modified mice and rats to our understanding of the cardiovascular pharmacology of serotonin. Pharmacol. Ther. 2010, 128, 559–567. [Google Scholar] [CrossRef]
- Saura, J.; Richards, J.G.; Mahy, N. Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Neurobiol. Aging 1994, 15, 399–408. [Google Scholar] [CrossRef]
- Strolin Benedetti, M.; Thomassin, J.; Tocchetti, P.; Dostert, P.; Kettler, R.; Da Prada, M. Species differences in changes of heart monoamine oxidase activities with age. J. Neural. Transm. Suppl. 1994, 41, 83–87. [Google Scholar]
- Strolin Benedetti, M.; Dostert, P.; Tipton, K.F. Developmental aspects of the monoamine-degrading enzyme monoamine oxidase. Dev. Pharmacol. Ther. 1992, 18, 191–200. [Google Scholar] [CrossRef]
- Ohki, R.; Yamamoto, K.; Ueno, S.; Mano, H.; Ikeda, U.; Shimada, K. Effects of olmesartan, an angiotensin II receptor blocker, on mechanically-modulated genes in cardiac myocytes. Cardiovasc. Drugs Ther. 2003, 17, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.J.; Saura, J.; Billett, E.E.; Finch, C.C.; Mahy, N. Cellular localization of monoamine oxidase A and B in human tissues outside of the central nervous system. Cell Tissue Res. 2001, 304, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramaniam, S.D.; Finch, C.C.; Rodriguez, M.J.; Mahy, N.; Billett, E.E. A comparative study of the expression of monoamine oxidase-A and -B mRNA and protein in non-CNS human tissues. Cell Tissue Res. 2003, 313, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Meco, M.; Bonifati, V.; Collier, W.L.; Ramacci, M.T.; Amenta, F. Enzyme histochemistry of monoamine oxidase in the heart of aged rats. Mech. Ageing Dev. 1987, 38, 145–155. [Google Scholar] [CrossRef]
- Robinson, D.S.; Davis, J.M.; Nies, A.; Ravaris, C.L.; Sylwester, D. Relation of sex and aging to monoamine oxidase activity of human brain, plasma, and platelets. Arch. Gen. Psychiatry 1971, 24, 536–539. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, K.; Shih, J.C.; Teng, C.T. Estrogen-related receptors-stimulated monoamine oxidase B promoter activity is down-regulated by estrogen receptors. Mol. Endocrinol. 2006, 20, 1547–1561. [Google Scholar] [CrossRef]
- Costiniti, V.; Spera, I.; Menabo, R.; Palmieri, E.M.; Menga, A.; Scarcia, P.; Porcelli, V.; Gissi, R.; Castegna, A.; Canton, M. Monoamine oxidase-dependent histamine catabolism accounts for post-ischemic cardiac redox imbalance and injury. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864 Pt B, 3050–3059. [Google Scholar] [CrossRef]
- Maintz, L.; Novak, N. Histamine and histamine intolerance. Am. J. Clin. Nutr. 2007, 85, 1185–1196. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural. Transm. (Vienna) 2018, 125, 1627–1634. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Bianchi, P.; Kunduzova, O.; Parini, A. New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J. Neural. Transm. (Vienna) 2007, 114, 823–827. [Google Scholar] [CrossRef]
- Holschneider, D.P.; Scremin, O.U.; Roos, K.P.; Chialvo, D.R.; Chen, K.; Shih, J.C. Increased baroreceptor response in mice deficient in monoamine oxidase A and B. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H964–H972. [Google Scholar] [CrossRef] [PubMed]
- Lairez, O.; Calise, D.; Bianchi, P.; Ordener, C.; Spreux-Varoquaux, O.; Guilbeau-Frugier, C.; Escourrou, G.; Seif, I.; Roncalli, J.; Pizzinat, N.; et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J. Mol. Cell. Cardiol. 2009, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Grobe, J.M.; Weisgut, J.; Schwelberger, H.G.; Fogel, W.A.; Marusakova, M.; Wache, H.; Bahre, H.; Buchwalow, I.B.; Dhein, S.; et al. Histamine can be Formed and Degraded in the Human and Mouse Heart. Front. Pharmacol. 2021, 12, 582916. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef]
- Maurel, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Frances, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1460–H1467. [Google Scholar] [CrossRef]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox Signal. 2013, 18, 5–18. [Google Scholar] [CrossRef]
- Tanijiri, H. Cardiac hypertrophy in spontaneously hypertensive rats. Jpn. Heart J. 1975, 16, 174–188. [Google Scholar] [CrossRef]
- Pino, R.; Failli, P.; Mazzetti, L.; Buffoni, F. Monoamine oxidase and semicarbazide-sensitive amine oxidase activities in isolated cardiomyocytes of spontaneously hypertensive rats. Biochem. Mol. Med. 1997, 62, 188–196. [Google Scholar] [CrossRef] [PubMed]
- van Eif, V.W.; Bogaards, S.J.; van der Laarse, W.J. Intrinsic cardiac adrenergic (ICA) cell density and MAO-A activity in failing rat hearts. J. Muscle Res. Cell Motil. 2014, 35, 47–53. [Google Scholar] [CrossRef]
- Sun, X.Q.; Peters, E.L.; Schalij, I.; Axelsen, J.B.; Andersen, S.; Kurakula, K.; Gomez-Puerto, M.C.; Szulcek, R.; Pan, X.; da Silva Goncalves Bos, D.; et al. Increased MAO-A Activity Promotes Progression of Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 331–343. [Google Scholar] [CrossRef]
- Strom, C.C.; Kruhoffer, M.; Knudsen, S.; Stensgaard-Hansen, F.; Jonassen, T.E.; Orntoft, T.F.; Haunso, S.; Sheikh, S.P. Identification of a core set of genes that signifies pathways underlying cardiac hypertrophy. Comp. Funct. Genom. 2004, 5, 459–470. [Google Scholar] [CrossRef]
- Sturza, A.; Duicu, O.M.; Vaduva, A.; Danila, M.D.; Noveanu, L.; Varro, A.; Muntean, D.M. Monoamine oxidases are novel sources of cardiovascular oxidative stress in experimental diabetes. Can. J. Physiol. Pharmacol. 2015, 93, 555–561. [Google Scholar] [CrossRef]
- Manni, M.E.; Zazzeri, M.; Musilli, C.; Bigagli, E.; Lodovici, M.; Raimondi, L. Exposure of cardiomyocytes to angiotensin II induces over-activation of monoamine oxidase type A: Implications in heart failure. Eur. J. Pharmacol. 2013, 718, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Forkink, M.; Hu, C.H.; Buonincontri, G.; Antonucci, S.; Di Sante, M.; Murphy, M.P.; Paolocci, N.; Mochly-Rosen, D.; Krieg, T.; et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. 2018, 25, 1671–1685. [Google Scholar] [CrossRef]
- Umbarkar, P.; Singh, S.; Arkat, S.; Bodhankar, S.L.; Lohidasan, S.; Sitasawad, S.L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic. Biol. Med. 2015, 87, 263–273. [Google Scholar] [CrossRef]
- Jin, H.; Yang, R.; Awad, T.A.; Wang, F.; Li, W.; Williams, S.P.; Ogasawara, A.; Shimada, B.; Williams, P.M.; de Feo, G.; et al. Effects of early angiotensin-converting enzyme inhibition on cardiac gene expression after acute myocardial infarction. Circulation 2001, 103, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.W.; Bodyak, N.; Yue, P.; Liu, Z.; Brown, J.; Izumo, S.; Kang, P.M. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol. Genom. 2005, 21, 34–42. [Google Scholar] [CrossRef]
- Petrak, J.; Pospisilova, J.; Sedinova, M.; Jedelsky, P.; Lorkova, L.; Vit, O.; Kolar, M.; Strnad, H.; Benes, J.; Sedmera, D.; et al. Proteomic and transcriptomic analysis of heart failure due to volume overload in a rat aorto-caval fistula model provides support for new potential therapeutic targets—monoamine oxidase A and transglutaminase 2. Proteome Sci. 2011, 9, 69. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Rieder, M.; Gauchel, N.; Bode, C.; Duerschmied, D. Serotonin: A platelet hormone modulating cardiovascular disease. J. Thromb. Thrombolysis 2021, 52, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Nigmatullina, R.R.; Kirillova, V.V.; Jourjikiya, R.K.; Mukhamedyarov, M.A.; Kudrin, V.S.; Klodt, P.M.; Palotas, A. Disrupted serotonergic and sympathoadrenal systems in patients with chronic heart failure may serve as new therapeutic targets and novel biomarkers to assess severity, progression and response to treatment. Cardiology 2009, 113, 277–286. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Hu, J.; Li, T.; Ma, X.; Meng, J.; Jia, M.; Lu, J.; Ohtsu, H.; Chen, Z.; Luo, X. Arrhythmogenic effect of sympathetic histamine in mouse hearts subjected to acute ischemia. Mol. Med. 2012, 18, 1–9. [Google Scholar] [CrossRef]
- Genovese, A.; Spadaro, G. Highlights in cardiovascular effects of histamine and H1-receptor antagonists. Allergy 1997, 52 (Suppl. S34), 67–78. [Google Scholar] [CrossRef]
- He, Z.; Ma, C.; Yu, T.; Song, J.; Leng, J.; Gu, X.; Li, J. Activation mechanisms and multifaceted effects of mast cells in ischemia reperfusion injury. Exp. Cell Res. 2019, 376, 227–235. [Google Scholar] [CrossRef]
- Gergs, U.; Jung, F.; Buchwalow, I.B.; Hofmann, B.; Simm, A.; Treede, H.; Neumann, J. Pharmacological and physiological assessment of serotonin formation and degradation in isolated preparations from mouse and human hearts. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1087–H1097. [Google Scholar] [CrossRef]
- Ponicke, K.; Gergs, U.; Buchwalow, I.B.; Hauptmann, S.; Neumann, J. On the presence of serotonin in mammalian cardiomyocytes. Mol. Cell. Biochem. 2012, 365, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.; Mo, J.; Tan, X.; Wang, J.; Yan, Z.; Liu, Y. Yin Yang 1 (YY1)-induced long intergenic non-protein coding RNA 472 (LINC00472) aggravates sepsis-associated cardiac dysfunction via the micro-RNA-335-3p (miR-335-3p)/Monoamine oxidase A (MAOA) cascade. Bioengineered 2022, 13, 1049–1061. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid. Redox Signal. 2014, 20, 267–280. [Google Scholar] [CrossRef]
- Fischer, Y.; Thomas, J.; Kamp, J.; Jungling, E.; Rose, H.; Carpéné, C.; Kammermeier, H. 5-hydroxytryptamine stimulates glucose transport in cardiomyocytes via a monoamine oxidase-dependent reaction. Biochem. J. 1995, 311 Pt 2, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar]
- Gibb, A.A.; Lorkiewicz, P.K.; Zheng, Y.T.; Zhang, X.; Bhatnagar, A.; Jones, S.P.; Hill, B.G. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem. J. 2017, 474, 2785–2801. [Google Scholar] [CrossRef]
- Bianchi, P.; Pimentel, D.R.; Murphy, M.P.; Colucci, W.S.; Parini, A. A new hypertrophic mechanism of serotonin in cardiac myocytes: Receptor-independent ROS generation. FASEB J. 2005, 19, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Shu, S.; Liu, R.; Jiang, Y.; Zhang, W.; Men, H. Monoamine oxidase inhibitors protect against coronary heart disease in rodent rat models: A pilot study. Pak. J. Pharm. Sci. 2019, 32, 371–375. [Google Scholar] [PubMed]
- Villeneuve, C.; Caudrillier, A.; Ordener, C.; Pizzinat, N.; Parini, A.; Mialet-Perez, J. Dose-dependent activation of distinct hypertrophic pathways by serotonin in cardiac cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H821–H828. [Google Scholar] [CrossRef]
- Lairez, O.; Cognet, T.; Schaak, S.; Calise, D.; Guilbeau-Frugier, C.; Parini, A.; Mialet-Perez, J. Role of serotonin 5-HT2A receptors in the development of cardiac hypertrophy in response to aortic constriction in mice. J. Neural. Transm. (Vienna) 2013, 120, 927–935. [Google Scholar] [CrossRef]
- Sommer, N.; Schulz, R. Mitochondrial Monoamine Oxidase: Another Player in Pulmonary Hypertension? Am. J. Respir. Cell Mol. Biol. 2021, 64, 277–278. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef]
- Sonobe, T.; Akiyama, T.; Du, C.K.; Pearson, J.T. Serotonin uptake via plasma membrane monoamine transporter during myocardial ischemia-reperfusion in the rat heart in vivo. Physiol. Rep. 2019, 7, e14297. [Google Scholar] [CrossRef]
- Du, C.K.; Zhan, D.Y.; Akiyama, T.; Inagaki, T.; Shishido, T.; Shirai, M.; Pearson, J.T. Myocardial interstitial levels of serotonin and its major metabolite 5-hydroxyindole acetic acid during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H60–H67. [Google Scholar] [CrossRef]
- Inagaki, T.; Akiyama, T.; Du, C.K.; Zhan, D.Y.; Yoshimoto, M.; Shirai, M. Monoamine oxidase-induced hydroxyl radical production and cardiomyocyte injury during myocardial ischemia-reperfusion in rats. Free Radic. Res. 2016, 50, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Hirschhauser, C.; Bornbaum, J.; Sydykov, A.; Dempfle, A.; Schneider, A.; Braun, T.; Schluter, K.D.; Schulz, R. Cardiomyocytes-specific deletion of monoamine oxidase B reduces irreversible myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.; Kunduzova, O.; Masini, E.; Cambon, C.; Bani, D.; Raimondi, L.; Seguelas, M.H.; Nistri, S.; Colucci, W.; Leducq, N.; et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 2005, 112, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Danila, M.D.; Privistirescu, A.I.; Mirica, S.N.; Sturza, A.; Ordodi, V.; Noveanu, L.; Duicu, O.M.; Muntean, D.M. Acute inhibition of monoamine oxidase and ischemic preconditioning in isolated rat hearts: Interference with postischemic functional recovery but no effect on infarct size reduction. Can. J. Physiol. Pharmacol. 2015, 93, 819–825. [Google Scholar] [CrossRef]
- Varela, A.; Mavroidis, M.; Katsimpoulas, M.; Sfiroera, I.; Kappa, N.; Mesa, A.; Kostomitsopoulos, N.G.; Cokkinos, D.V. The neuroprotective agent Rasagiline mesylate attenuates cardiac remodeling after experimental myocardial infarction. ESC Heart Fail. 2017, 4, 331–340. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Xu, B.; Bahriz, S.M.F.; Zhu, C.; Jovanovic, A.; Ni, H.; Jacobi, A.; Kaludercic, N.; Di Lisa, F.; et al. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular beta(1)AR signaling to calibrate cardiac contractile function. Basic Res. Cardiol. 2022, 117, 37. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Shi, Q.; Xu, B.; Zhu, C.; Li, M.; Mir, V.; Bers, D.M.; Xiang, Y.K. Monoamine Oxidases Desensitize Intracellular beta(1)AR Signaling in Heart Failure. Circ. Res. 2021, 129, 965–967. [Google Scholar] [CrossRef]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid. Redox Signal. 2016, 25, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Martini, H.; Lefevre, L.; Sayir, S.; Itier, R.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Pizzinat, N.; Cussac, D.; et al. Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. Int. J. Mol. Sci. 2021, 22, 2245. [Google Scholar] [CrossRef]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabo, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 531–550. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, R.; Schlüter, K.-D. Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases. Int. J. Mol. Sci. 2023, 24, 6459. https://doi.org/10.3390/ijms24076459
Schulz R, Schlüter K-D. Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases. International Journal of Molecular Sciences. 2023; 24(7):6459. https://doi.org/10.3390/ijms24076459
Chicago/Turabian StyleSchulz, Rainer, and Klaus-Dieter Schlüter. 2023. "Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases" International Journal of Molecular Sciences 24, no. 7: 6459. https://doi.org/10.3390/ijms24076459
APA StyleSchulz, R., & Schlüter, K.-D. (2023). Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases. International Journal of Molecular Sciences, 24(7), 6459. https://doi.org/10.3390/ijms24076459