
CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Targeted Gene Correction of SLC12A3 Mutation with the CRISPR/Cas9 System
2.2. Establishment of GIT Patientcorr hiPSCs
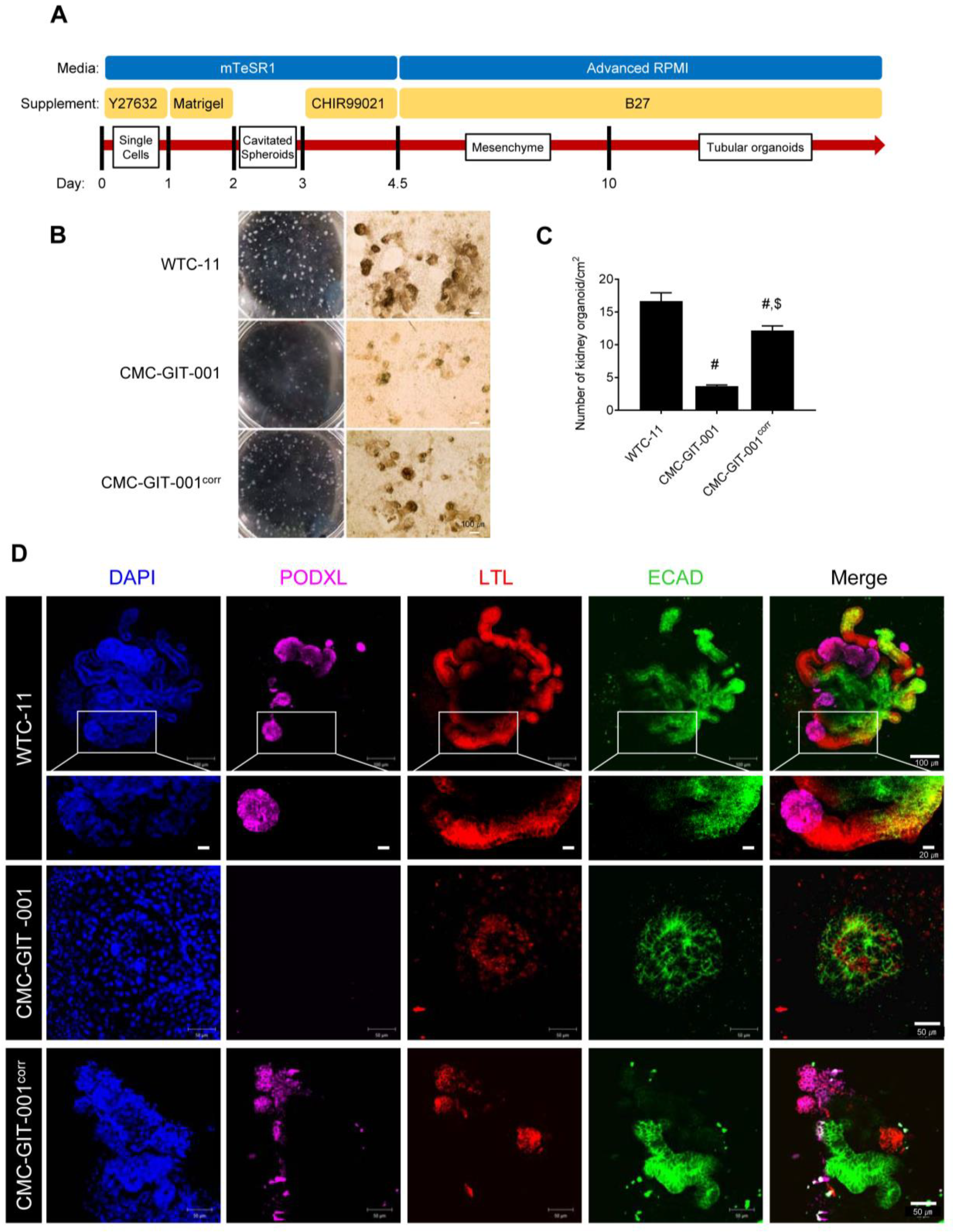
2.3. Differentiation of Corrected CMC-GIT-001 into Kidney Organoids
2.4. Recovery of NCCT Expression in Kidney Organoids from CMC-GIT-001corr hiPSCs
3. Discussion
4. Methods and Methods
4.1. hiPSC Generation and Cell Culture
4.2. Construction of sgRNA and ssODN
4.3. In Vitro Cas9 Cleavage Assay
4.4. hiPSC Transfection for Gene Editing
4.5. Flow Cytometry
4.6. Immunofluorescence
4.7. Tri-Lineage Differentiation
4.8. Karyotype Analysis
4.9. Mycoplasma Detection
4.10. Kidney Organoid Differentiation from hiPSCs
4.11. qRT-PCR
4.12. Immunoblot Analysis
4.13. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calo, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.; Konrad, M.; et al. Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Subramanya, A.R.; Ellison, D.H. Distal convoluted tubule. Clin. J. Am. Soc. Nephrol. 2014, 9, 2147–2163. [Google Scholar] [CrossRef] [PubMed]
- Gitelman, H.J.; Graham, J.B.; Welt, L.G. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans. Assoc. Am. Physicians 1966, 79, 221–235. [Google Scholar] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Miyagi, A.; Lu, A.; Humphreys, B.D. Gene Editing: Powerful New Tools for Nephrology Research and Therapy. J. Am. Soc. Nephrol. 2016, 27, 2940–2947. [Google Scholar] [CrossRef]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.W.; Shin, Y.J.; Cui, S.; Ko, E.J.; Lee, K.I.; Lee, J.Y.; Chung, B.H.; Yang, C.W. Generation of a human induced pluripotent stem cell line (CMCi002-A) from a patient with Gitelman’s syndrome. Stem Cell Res. 2020, 49, 102110. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef]
- Freedman, B.S. Modeling Kidney Disease with iPS Cells. Biomark. Insights 2015, 10, 153–169. [Google Scholar] [CrossRef]
- Liu, E.; Radmanesh, B.; Chung, B.H.; Donnan, M.D.; Yi, D.; Dadi, A.; Smith, K.D.; Himmelfarb, J.; Li, M.; Freedman, B.S.; et al. Profiling APOL1 Nephropathy Risk Variants in Genome-Edited Kidney Organoids with Single-Cell Transcriptomics. Kidney360 2020, 1, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.M.; Kim, Y.K. Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.M.; Freedman, B.S. CRISPR Gene Editing in the Kidney. Am. J. Kidney Dis. 2018, 71, 874–883. [Google Scholar] [CrossRef]
- Tanigawa, S.; Islam, M.; Sharmin, S.; Naganuma, H.; Yoshimura, Y.; Haque, F.; Era, T.; Nakazato, H.; Nakanishi, K.; Sakuma, T.; et al. Organoids from Nephrotic Disease-Derived iPSCs Identify Impaired NEPHRIN Localization and Slit Diaphragm Formation in Kidney Podocytes. Stem Cell Rep. 2018, 11, 727–740. [Google Scholar] [CrossRef]
- Costanzo, L.S. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am. J. Physiol. 1985, 248, F527–F535. [Google Scholar] [CrossRef]
- Ellison, D.H. The thiazide-sensitive na-cl cotransporter and human disease: Reemergence of an old player. J. Am. Soc. Nephrol. 2003, 14, 538–540. [Google Scholar] [CrossRef]
- WareJoncas, Z.; Campbell, J.M.; Martinez-Galvez, G.; Gendron, W.A.C.; Barry, M.A.; Harris, P.C.; Sussman, C.R.; Ekker, S.C. Precision gene editing technology and applications in nephrology. Nat. Rev. Nephrol. 2018, 14, 663–677. [Google Scholar] [CrossRef]
- Son, J.S.; Park, C.Y.; Lee, G.; Park, J.Y.; Kim, H.J.; Kim, G.; Chi, K.Y.; Woo, D.H.; Han, C.; Kim, S.K.; et al. Therapeutic correction of hemophilia A using 2D endothelial cells and multicellular 3D organoids derived from CRISPR/Cas9-engineered patient iPSCs. Biomaterials 2022, 283, 121429. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Yi, L.; Hou, Z.; Chen, J.; Kou, X.; Zhao, Y.; Wang, H.; Sun, X.F.; Jiang, C.; et al. Naive Induced Pluripotent Stem Cells Generated From beta-Thalassemia Fibroblasts Allow Efficient Gene Correction With CRISPR/Cas9. Stem Cells Transl. Med. 2016, 5, 267. [Google Scholar] [CrossRef] [PubMed]
- Sladen, P.E.; Perdigao, P.R.L.; Salsbury, G.; Novoselova, T.; van der Spuy, J.; Chapple, J.P.; Yu-Wai-Man, P.; Cheetham, M.E. CRISPR-Cas9 correction of OPA1 c.1334G>A: P.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Mol. Ther. Nucleic Acids 2021, 26, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e924. [Google Scholar] [CrossRef]
- Loffing, J.; Vallon, V.; Loffing-Cueni, D.; Aregger, F.; Richter, K.; Pietri, L.; Bloch-Faure, M.; Hoenderop, J.G.; Shull, G.E.; Meneton, P.; et al. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman’s syndrome. J. Am. Soc. Nephrol. 2004, 15, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Chen, J.; Yu, M.; Lin, Z.; Nan, X.; Dong, B.; Fang, X.; Chen, J.; Ding, G.; Zhang, A.; et al. Multi-centre study of the clinical features and gene variant spectrum of Gitelman syndrome in Chinese children. Clin. Genet. 2021, 99, 558–564. [Google Scholar] [CrossRef]
- Vargas-Poussou, R.; Dahan, K.; Kahila, D.; Venisse, A.; Riveira-Munoz, E.; Debaix, H.; Grisart, B.; Bridoux, F.; Unwin, R.; Moulin, B.; et al. Spectrum of mutations in Gitelman syndrome. J. Am. Soc. Nephrol. 2011, 22, 693–703. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; de Baaij, J.H.F. The genetic spectrum of Gitelman(-like) syndromes. Curr. Opin. Nephrol. Hypertens. 2022, 31, 508–515. [Google Scholar] [CrossRef]
- Nunez-Gonzalez, L.; Carrera, N.; Garcia-Gonzalez, M.A. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Bartter and Gitelman Syndromes: A Primer for Clinicians. Int. J. Mol. Sci. 2021, 22, 11414. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; et al. Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res. 2020, 48, 130–140. [Google Scholar] [CrossRef]
- Do, H.S.; Park, S.W.; Im, I.; Seo, D.; Yoo, H.W.; Go, H.; Kim, Y.H.; Koh, G.Y.; Lee, B.H.; Han, Y.M. Enhanced thrombospondin-1 causes dysfunction of vascular endothelial cells derived from Fabry disease-induced pluripotent stem cells. EBioMedicine 2020, 52, 102633. [Google Scholar] [CrossRef]
- Bandara, R.A.; Chen, Z.R.; Hu, J. Potential of helper-dependent Adenoviral vectors in CRISPR-cas9-mediated lung gene therapy. Cell Biosci. 2021, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Gu, L.; Wang, J.; Chang, Y.; Jin, M.; Mao, Y.; Wang, H.; Ji, G. A Novel System for Simple Rapid Adenoviral Vector Construction to Facilitate CRISPR/Cas9-Mediated Genome Editing. CRISPR J. 2021, 4, 381–391. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.W.; Fang, X.; Cui, S.; Lee, H.; Shin, Y.J.; Ko, E.J.; Lee, K.I.; Lee, J.Y.; Chung, B.H.; Yang, C.W. CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System. Int. J. Mol. Sci. 2023, 24, 3019. https://doi.org/10.3390/ijms24033019
Lim SW, Fang X, Cui S, Lee H, Shin YJ, Ko EJ, Lee KI, Lee JY, Chung BH, Yang CW. CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System. International Journal of Molecular Sciences. 2023; 24(3):3019. https://doi.org/10.3390/ijms24033019
Chicago/Turabian StyleLim, Sun Woo, Xianying Fang, Sheng Cui, Hanbi Lee, Yoo Jin Shin, Eun Jeong Ko, Kang In Lee, Jae Young Lee, Byung Ha Chung, and Chul Woo Yang. 2023. "CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System" International Journal of Molecular Sciences 24, no. 3: 3019. https://doi.org/10.3390/ijms24033019
APA StyleLim, S. W., Fang, X., Cui, S., Lee, H., Shin, Y. J., Ko, E. J., Lee, K. I., Lee, J. Y., Chung, B. H., & Yang, C. W. (2023). CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System. International Journal of Molecular Sciences, 24(3), 3019. https://doi.org/10.3390/ijms24033019