
Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis




Abstract
1. Introduction
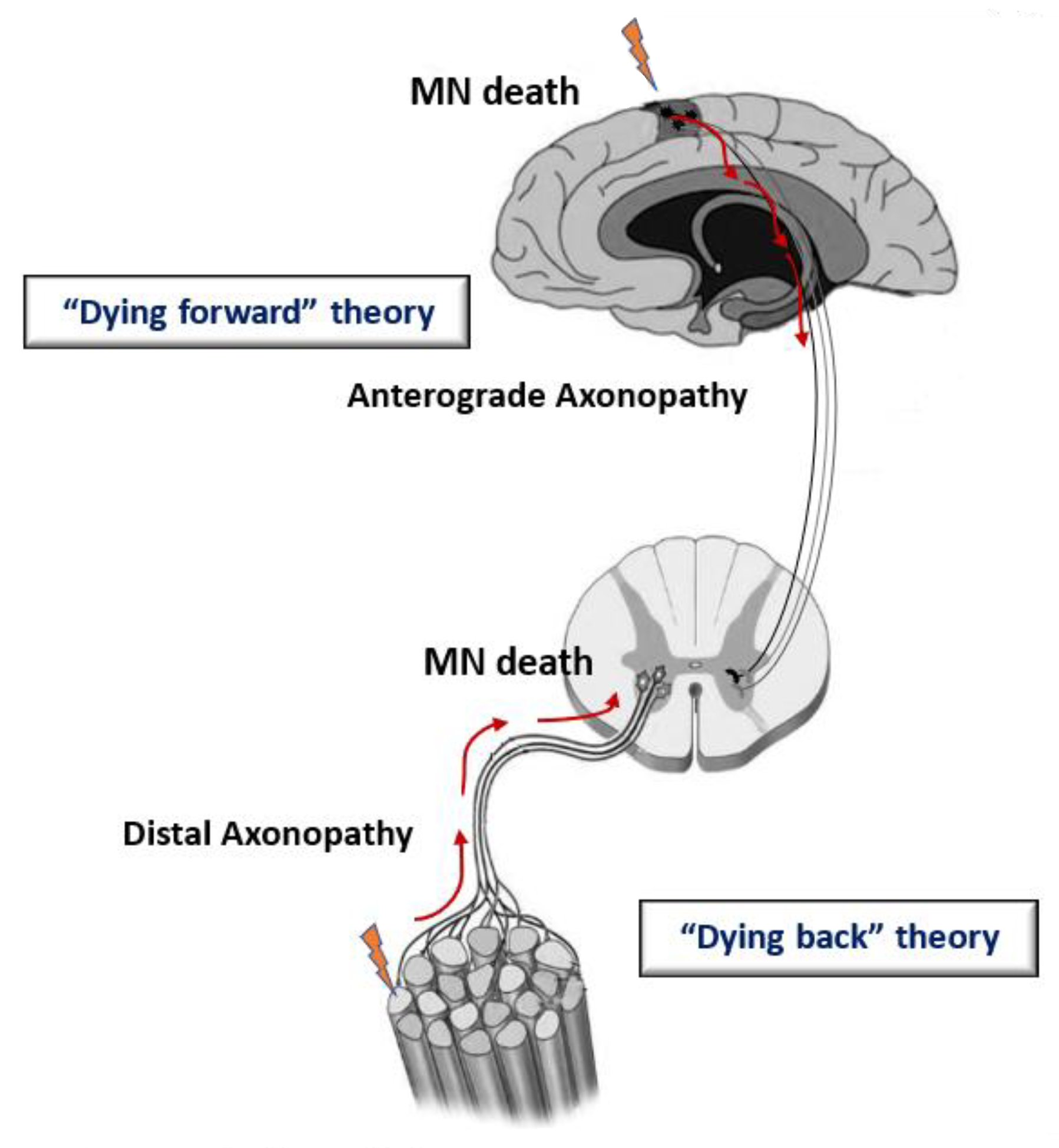
2. Etiology and Physiopathology of ALS
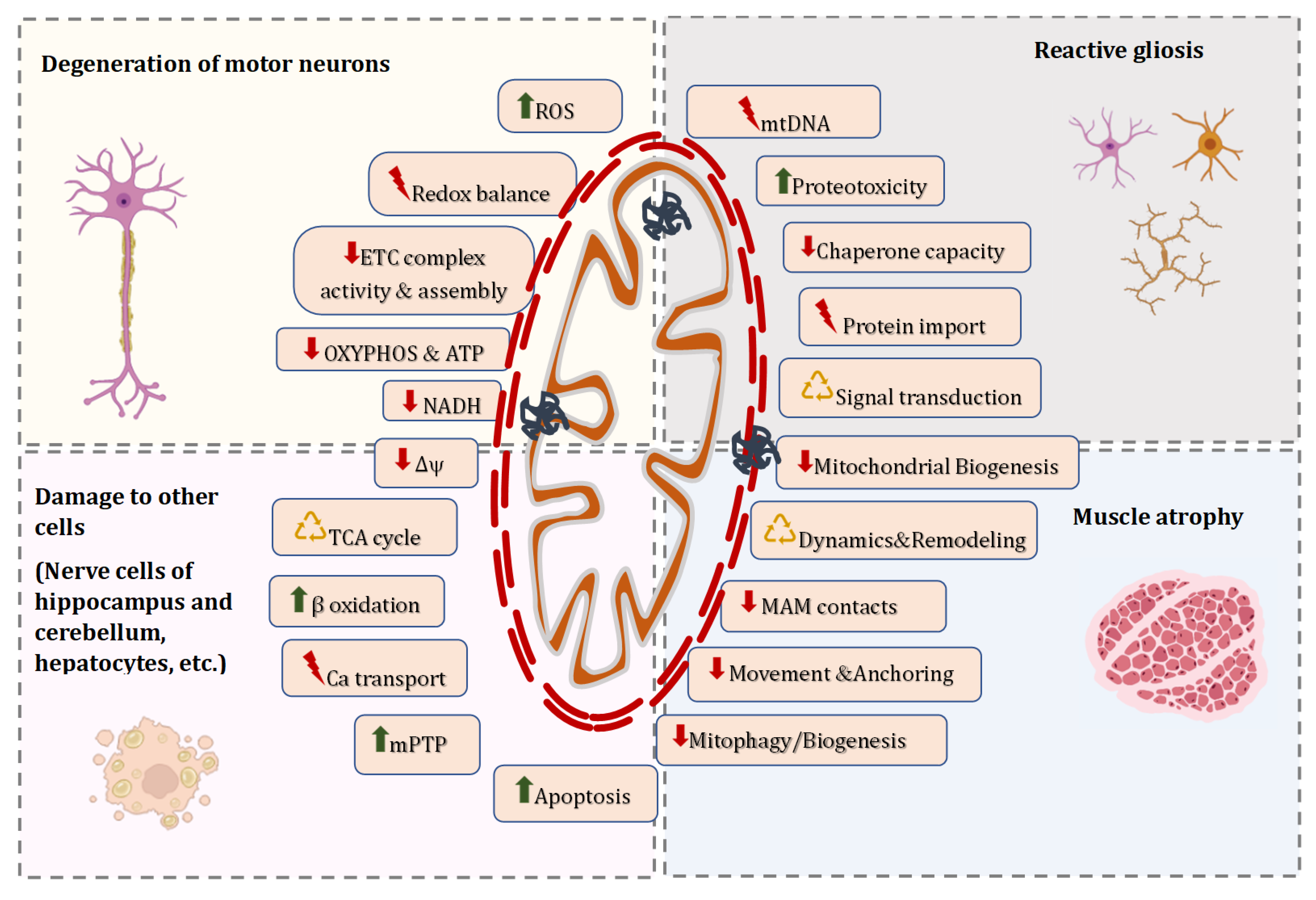
3. Mitochondrial Malfunction and Dyshomeostasis in Patients and Genetic Models of ALS

{kind=link}
{kind=link}
| Disease-Causing Mutations | Most Common Disturbances in Mitochondrial Homeostasis | Encoding Proteins and Their Function | Commonly Used In Vivo or In Vitro Models | Ref. |
|---|---|---|---|---|
| In vivo Models | ||||
| SOD1 point mutations and variants | Disruption of the cristae and disintegration of the mitochondrial structure; Vacuolar degeneration; Dysfunction of OXYPHOS; Mitochondrial Ca2+ uptake disorders; Mitochondria distribution impairment; Reduced mitochondrial density | Cu/Zn superoxide dismutase 1 (SOD1) protein catalyzes the dismutation of the superoxide radical into hydrogen peroxide and molecular oxygen; it plays a role in the cellular homeostasis of ROS. | Cu/Zn superoxide dismutase 1 (SOD1)-G93A mouse models | [23,27,57,58,59,60,61] |
| G93R-mSOD1 zebrafish model | [8,59,60,61] | |||
| A GGGGCC24+ hexanucleotide repeat expansion (HRE) in the C9ORF72 gene | Defective mitochondrial bioenergetic function; Mitochondrial fragmentation; Reduced protein level of the ATP synthase complex | The function of C9ORF72 remains unknown, but it has been suggested that it plays a role in protein trafficking | FVB-C9orf72 mouse model;Rodent models of C9orf72;C9orf72 knockdown zebrafish model | [62,63] |
| TARDBP point mutations and variants | Abnormal mitochondrial morphology and motility | TAR DNA-binding protein 43 (TDP-43) protein participates in transcriptional regulation through RNA/DNA and protein–protein interactions, RNA processing and splicing regulation | TDP43-Q331K mouse model; TDP-43G298S mice (over 20 mouse models of TDP-43); TDP43-A315T zebrafish model | [64,65,66,67] |
| FUS and TBK1 variants | Mitochondrial fragmentation; Damaged mitochondrial cristae; Hyperfused and elongated mitochondria | Fused in sarcoma (FUS) protein is a DNA/RNA-binding protein that participates in in DNA damage, mRNA splicing, transport, transcription, and translation. | Drosophila models of FUS-related ALS; hFUSWT mice; FUS(1-359) mice | [68,69,70,71,72,73] |
| Tbk1fl/flNestin-Cre mice | [74,75,76] | |||
| In vitro models | ||||
| Mutations in SOD1, FUS, TARDBP, C9orf72, SFPQ, and others | Disorders in mitochondrial morphology and movement; Impairment of mitochondrial calcium buffering; Disruption of endoplasmic reticulum (ER)-mitochondria tethering and signaling | Human induced pluripotent stem cells (hiPSCs)-derived motor neurons, astrocytes, and microglia from patients with SOD1, FUS, TARDBP, C9orf72, and other mutations | [77,78,79,80,81] | |
| Mutations in SOD1, FUS, KIF5A, PFN1, and others | Altered mitochondrial shape and size; Reduced mitochondrial movement; Matrix swelling and other structural defects in mitochondria; Decreased mitochondrial respiration; Decrease in the number of mitochondria. | Cultured murine primary neurons co-transfected with mutant fALS-related proteins; human cells expressing mutant derivatives | [82,83,84,85] | |
4. Molecular Mechanisms Responsible for Altered Mitostasis in ALS
4.1. Alterations in Morphology of Individual Mitochondria and Dynamics of the Mitochondrial Network in ALS
4.2. Functional Signs of Mitochondrial Damage in ALS
4.3. Modifications in the Mitochondrial Genome in ALS
4.4. Disorders in Mitochondrial Clearance and Replacement in ALS
4.5. Defects in Mitochondrial Trafficking in ALS
5. Strategies of Targeted Mitochondrial Therapy for ALS
5.1. Mitochondria-Specific Form of Autophagy as a Potential Target for ALS Treatment
5.2. PGC-1α as a Drug Target for ALS Treatment
5.3. Mitochondria-Targeted Antioxidants
5.4. Mitochondrial Biogenetics and Energy Metabolism as Targets of Therapeutic High-Fat Diet in ALS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O. Management of respiratory symptoms in ALS. J. Neurol. 2011, 258, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Mukhamedyarov, M.A.; Khabibrakhmanov, A.N.; Zefirov, A.L. Early dysfunctions in amyotrophic lateral sclerosis: Pathogenetic mechanisms and role in the initiation of the disease. Biochem. Suppl. Ser. A Membr. Cell Biol. 2020, 14, 261–266. [Google Scholar] [CrossRef]
- Boillee, S.; Velde, C.V.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, K.; Youssef, M.; You, J.; Sung, H.-K.; Park, J. Evidence of metabolic dysfunction in amyotrophic lateral sclerosis (ALS) patients and animal models. Biomolecules 2023, 13, 863. [Google Scholar] [CrossRef]
- Sirozh, O.; Saez-Mas, A.; Lafarga, V.; Fernandez-Capetillo, O. Basic concepts and emergent disease mechanisms of amyotrophic lateral sclerosis. Encycl. Cell Biol. 2023, 6, 644–665. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]
- Gois, A.M.; Mendonça, D.M.F.; Freire, M.A.M.; Santos, J.R. In vitro and in vivo models of amyotrophic lateral sclerosis: An updated overview. Brain Res. Bull. 2020, 159, 32–43. [Google Scholar] [CrossRef]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; López de Munain, A. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Progr. Neurobiol. 2016, 142, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Lunetta, C.; Vourc’h, P.; Pradat, P.F.; Blasco, H. Time for optimism in amyotrophic lateral sclerosis. Eur. J. Neurol. 2023, 30, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Jishi, A.; Qi, X. Altered mitochondrial protein homeostasis and proteinopathies. Front. Mol. Neurosci. 2022, 15, 867935. [Google Scholar] [CrossRef] [PubMed]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Lim, S.M.; Nahm, M.; Kim, S.H. Proteostasis and ribostasis impairment as common cell death mechanisms in neurodegenerative diseases. J. Clin. Neurol. 2023, 19, 101–114. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, J. Mitochondrial quality control and neurodegenerative diseases. Neuronal Signal. 2018, 2, NS20180062. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Granatiero, V.; Manfredi, G. Mitochondrial transport and turnover in the pathogenesis of amyotrophic lateral sclerosis. Biology 2019, 8, 36. [Google Scholar] [CrossRef]
- Günther, R.; Pal, A.; Williams, C.; Zimyanin, V.L.; Liehr, M.; von Neubeck, C.; Krause, M.; Parab, M.G.; Petri, S.; Kalmbach, N.; et al. Alteration of mitochondrial integrity as upstream event in the pathophysiology of SOD1-ALS. Cells 2022, 11, 1246. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: Prevention by mitochondrial-targeted antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflamm. 2019, 16, 230. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Mehta, A.R.; Walters, R.; Waldron, F.M.; Pal, S.; Selvaraj, B.T.; Macleod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Brain Commun. 2019, 1, fcz009. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The impact of mitochondrial dysfunction in amyotrophic lateral sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Laneve, P.; Tollis, P.; Caffarelli, E. RNA deregulation in amyotrophic lateral sclerosis: The noncoding perspective. Int. J. Mol. Sci. 2021, 22, 20285. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Tiloca, C.; Morelli, C.; Colombrita, C.; Poletti, B.; Doretti, A.; Maderna, L.; Messina, S.; Ratti, A.; Silani, V. Genetics of familial amyotrophic lateral sclerosis. Arch. Ital. Biol. 2011, 149, 65–82. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in the C9ORF72 is the cause of the chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ASL and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Rad. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Newell, M.E.; Adhikari, S.; Halden, R.U. Systematic and state-of the science review of the role of environmental factors in Amyotrophic Lateral Sclerosis (ALS) or Lou Gehrig’s Disease. Sci. Total Environ. 2022, 817, 152504. [Google Scholar] [CrossRef]
- Bradley, W.G.; Borenstein, A.R.; Nelson, L.M.; Codd, G.A.; Rosen, B.H.; Stommel, E.W.; Cox, P.A. Is exposure to cyanobacteria an environmental risk factor for amyotrophic lateral sclerosis and other neurodegenerative diseases? Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antivir. Res. 2013, 99, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current state and future directions in the diagnosis of amyotrophic lateral sclerosis. Cells 2023, 12, 736. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Dupuis, L.; Loeffler, J.P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: Insights from transgenic models. Curr. Opin. Pharmacol. 2009, 9, 341–346. [Google Scholar] [CrossRef]
- Eisen, A. The dying forward hypothesis of ALS: Tracing its history. Brain Sci. 2021, 11, 300. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dyingback” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The skeletal muscle emerges as a new disease target in amyotrophic lateral sclerosis. J. Pers. Med. 2021, 11, 671. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Herrando-Grabulosa, M.; Navarro, X. Sensory involvement in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2022, 23, 15521. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. Mitoph Aging: Mitophagy in aging and disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the trip: Calcium in mitochondria back and forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Hayes, L.R.; Asress, S.A.; Li, Y.; Galkin, A.; Stepanova, A.; Kawamata, H.; Manfredi, G.; Glass, J.D. Distal denervation in the SOD1 knockout mouse correlates with loss of mitochondria at the motor nerve terminal. Exp. Neurol. 2019, 318, 251–257. [Google Scholar] [CrossRef]
- Lemmens, R.; Van Hoecke, A.; Hersmus, N.; Geelen, V.; D’Hollander, I.; Thijs, V.; Van Den Bosch, L.; Carmeliet, P.; Robberecht, W. Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum. Mol. Genet. 2007, 16, 2359–2365. [Google Scholar] [CrossRef]
- Da Costa, M.M.J.; Allen, C.E.; Higginbottom, A.; Ramesh, T.; Shaw, P.J.; McDermott, C.J. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis. Model. Mech. 2014, 7, 73–81. [Google Scholar] [CrossRef]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Chiang, P.M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Shahidullah, M.; Le Marchand, S.J.; Fei, H.; Zhang, J.; Pandey, U.B.; Dalva, M.B.; Pasinelli, P.; Levitan, I.B. Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J. Neurosci. 2013, 33, 19590–19598. [Google Scholar] [CrossRef] [PubMed]
- Anoar, S.; Woodling, N.S.; Niccoli, T. Mitochondria dysfunction in frontotemporal dementia/Amyotrophic Lateral Sclerosis: Lessons from Drosophila models. Front. Neurosci. 2021, 15, 786076. [Google Scholar] [CrossRef]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Salam, S.; Tacconelli, S.; Smith, B.N.; Mitchell, J.C.; Glennon, E.; Nikolaou, N.; Houart, C.; Vance, C. Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 2021, 11, 13613. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, S.; Wang, J.; Wu, Q.; Wang, A.; Guan, H.; Zhang, Q.; Zhang, D.; Wang, X.; Song, H.; et al. TBK1-mediated DRP1 targeting confers nucleic acid sensing to reprogram mitochondrial dynamics and physiology. Mol. Cell 2020, 80, 810–827. [Google Scholar] [CrossRef] [PubMed]
- Harding, O.; Evans, C.S.; Ye, J.; Cheung, J.; Maniatis, T.; Holzbaur, E.L.F. ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025053118. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef]
- Widagdo, J.; Udagedara, S.; Bhembre, N.; Tan, J.Z.A.; Neureiter, L.; Huang, J.; Anggono, V.; Lee, M. Familial ALS-associated SFPQ variants promote the formation of SFPQ cytoplasmic aggregates in primary neurons. Open Biol. 2022, 12, 220187. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Du, H.; Huo, Z.; Chen, Y.; Zhao, Z.; Meng, F.; Wang, X.; Liu, S.; Zhang, H.; Zhou, F.; Liu, J.; et al. Induced pluripotent stem cells and their applications in amyotrophic lateral sclerosis. Cells 2023, 12, 971. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Ju, S.; Arjun, A.; Batarse, A.; Archbold, H.C.; Peisach, D.; Li, X.; Zhang, Y.; Tank, E.M.; Qiu, H.; et al. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl. Acad. Sci. USA 2015, 112, 7821–7826. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.M.; Fenton, A.R.; Saez-Atienzar, S.; Giampetruzzi, A.; Sreeram, A.; Shankaracharya; Keagle, P.J.; Doocy, V.R.; Smith, N.J.; Danielson, E.W.; et al. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 2022, 39, 10598. [Google Scholar] [CrossRef]
- Teyssou, E.; Chartier, L.; Roussel, D.; Perera, N.D.; Nemazanyy, I.; Langui, D.; Albert, M.; Larmonier, T.; Saker, S.; Salachas, F.; et al. The amyotrophic lateral sclerosis M114T PFN1 mutation deregulates alternative autophagy pathways and mitochondrial homeostasis. Int. J. Mol. Sci. 2022, 23, 5694. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Modeling environmentally-induced motor neuron degeneration in zebrafish. Sci. Rep. 2018, 8, 4890. [Google Scholar] [CrossRef] [PubMed]
- Tabata, R.C.; Wilson, J.M.; Ly, P.; Zwiegers, P.; Kwok, D.; Van Kampen, J.M.; Cashman, N.; Shaw, C.A. Chronic exposure to dietary sterol glucosides is neurotoxic to motor neurons and induces an ALS-PDC phenotype. Neuromol. Med. 2008, 10, 24–39. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, A.; Li, X.; Yi, J. Dysregulated mitochondrial Ca2+ and ROS signaling in skeletal muscle of ALS mouse model. Arch. Biochem. Biophys. 2019, 663, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Jemmerson, R.; Dubinsky, J.M.; Brustovetsky, N. Cytochrome c release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid. Redox Signal. 2005, 7, 1158–1173. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef]
- Li, Q.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16. [Google Scholar] [CrossRef]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Hadj, S.B.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Dong, K.; Bigio, E.H.; Özdinler, P.H. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.J.; Suh, Y.L. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL sumoylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, W.; Perry, G.; Zhu, X.; Wang, X. Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. EMBO Mol. Med. 2018, 10, e8888. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef]
- Magrane, J.; Sahawneh, M.A.; Przedborski, S.; Estevez, A.G.; Manfredi, G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 229–242. [Google Scholar] [CrossRef]
- Carri, M.T.; Ferri, A.; Battistoni, A.; Famhy, L.; Gabbianelli, R.; Poccia, F.; Rotilio, G. Expression of a Cu, Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997, 414, 365–368. [Google Scholar] [CrossRef]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Rizzardini, M.; Mangolini, A.; Lupi, M.; Ubezio, P.; Bendotti, C.; Cantoni, L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 2005, 232, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Kihira, T.; Okamoto, K.; Yoshida, S.; Kondo, T.; Iwai, K.; Wada, S.; Kajimoto, Y.; Kondo, T.; Kokubo, Y.; Kuzuhara, S. Environmental characteristics and oxidative stress of inhabitants and patients with amyotrophic lateral sclerosis in a high incidence area on the Kii Peninsula, Japan. Int. Med. 2013, 52, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Medard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Tanaka, H.; Takata, M.; Nagahara, Y.; Noda, Y.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Hara, H. SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci. Lett. 2014, 559, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; et al. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Mitochondriopathy mimicking amyotrophic lateral sclerosis. Neurologist 2003, 9, 45–48. [Google Scholar] [CrossRef]
- Jimenez-Pacheco, A.; Franco, J.M.; Lopez, S.; Gomez-Zumaquero, J.M.; Magdalena Leal-Lasarte, M.; Caballero-Hernandez, D.E.; Cejudo-Guillén, M.; Pozo, D. Epigenetic mechanisms of gene regulation in amyotrophic lateral sclerosis. Adv. Exp. Med. Biol. 2017, 978, 255–275. [Google Scholar] [CrossRef]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef]
- Maekawa, M.; Sugano, K.; Ushiama, M.; Fukayama, N.; Nomoto, K.; Kashiwabara, H.; Fujita, S.; Kakizoe, T. Heterogeneity of DNA methylation status analyzed by bisulfite-PCR-SSCP and correlation with clinico-pathological characteristics in colorectal cancer. Clin. Chem. Lab. Med. 2001, 39, 121–128. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, A.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferatoractivated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1α regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F.; Jang, Y.C.; Bhattacharya, A.; Bokov, A.F.; Jernigan, A.; Richardson, A.; Van Remmen, H. PGC-1α protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 2011, 44, 947–956. [Google Scholar] [CrossRef]
- Varghese, M.; Zhao, W.; Trageser, K.J.; Pasinetti, G.M. Peroxisome proliferator activator receptor gamma coactivator-1α overexpression in amyotrophic lateral sclerosis: A tale of two transgenics. Biomolecules 2020, 10, 760. [Google Scholar] [CrossRef]
- Ladd, A.C.; Keeney, P.M.; Govind, M.M.; Bennett, J.P., Jr. Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromol. Med. 2014, 16, 714–726. [Google Scholar] [CrossRef]
- Yang, X.; Pan, W.; Xu, G.; Chen, L. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta 2020, 502, 245–254. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 2020, 106, 789–805. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Le, W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008, 4, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy modulation, a new player in the race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012, 21, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, L.; Chang, J.; Cao, T.; Song, L.; Wen, C.; Chen, Y.; Zhuo, Y.; Chen, F. Safety and efficacy of edaravone in patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Clin. Drug Investig. 2023, 43, 1–11. [Google Scholar] [CrossRef]
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1G93A ALS mice. Neurotox. Res. 2017, 31, 230–244. [Google Scholar] [CrossRef]
- Li, X.; Dong, L.; Li, A.; Yi, J.; Brotto, M.; Zhou, J. Butyrate ameliorates mitochondrial respiratory capacity of the motor-neuron-like cell line NSC34-G93A, a cellular model for ALS. Biomolecules 2022, 12, 333. [Google Scholar] [CrossRef]
- Li, X.; Chen, C.; Zhan, X.; Li, B.; Zhang, Z.; Li, S.; Xie, Y.; Song, X.; Shen, Y.; Liu, J.; et al. R13 preserves motor performance in SOD1G93A mice by improving mitochondrial function. Theranostics 2021, 11, 7294–7307. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Thompson, J.L.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, T.; Sato, T.; Kanno, S.I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic acid 5 (MA-5) facilitates ATP synthase oligomerization and cell survival in various mitochondrial diseases. eBioMedicine 2017, 20, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Lipari, C.L.R.; Risiglione, P.; Zimbone, S.; Guarino, F.; Caccamo, A.; Messina, A. ERK1/2-dependent TSPO overactivation associates with the loss of mitophagy and mitochondrial respiration in ALS. Cell Death Dis. 2023, 14, 122. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L.; et al. Altered skeletal muscle glucose-fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa154. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Jain, S.S.; Paglialunga, S.; Vigna, C.; Ludzki, A.; Herbst, E.A.; Lally, J.S.; Schrauwen, P.; Hoeks, J.; Tupling, A.R.; Bonen, A.; et al. High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 2014, 63, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Harnessing polypharmacology with medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. [Google Scholar] [CrossRef] [PubMed]


| Pharmacological Agent | Molecular Targets | Therapeutic Effect | References |
|---|---|---|---|
| Pridopidine (PL-101) | Sigma-1 receptor agonist, MAM contacts, mitochondrial calcium homeostasis | Enhancement of bulbar and speech function in ALS patients | HEALEY ALS Platform Trial ID NCT04615923 |
| Nuedexta (Dextromethorphan and Quinidine) | Effectors of P450-containing systems in the inner membrane of mitochondria and in the endoplasmic reticulum | Improvement in bulbar function (speech, swallowing, and salivation) | ClinicalTrials.gov ID NCT01806857 |
| VAR10303 | Iron chelator, free radical catcher | Stimulation of mitochondrial biogenesis and an increase in animal life expectancy | [159] |
| Sodium butyrate | Broad-spectrum antioxidant | Stimulation of mitochondrial biogenesis and an increase in animal life expectancy | [160] |
| Compound R13 | Precursor of 7,8-dihydroxyflavone, selective activator of the TrkB signaling pathway | Activation of mitochondrial biogenesis, suppression of the development of mitochondrial dysfunction and an increase in animal life expectancy | [161] |
| Caprylic triglyceride | Substance metabolized into energy-rich ketone bodies | Restoration of energy metabolism, mitochondrial biogenesis and improvement of motor function in SOD1 mice | [162] |
| MitoQ | Mitochondria-targeted antioxidant | Improvement in mitochondrial function and neuroprotective effects in animal and cellular models of ALS | [26] |
| Cholest-4-en-3-one, oxime (TRO19622) | Inhibitor of the components of the mPTP opening: the voltage-dependent anion channel and the translocator protein | Delay in the onset of disease symptoms and increase in animal survival | [27] |
| Mito-CP | Mitochondria-targeted antioxidant | Improvement in mitochondrial function and neuroprotective effects in animal and cellular models of ALS | [28] |
| Elamipretide (SS-31) | Mitochondrion-targeted antioxidant | Improvement in mitochondrial dysfunction, synaptic and memory impairment | [29] |
| Coenzyme Q10 | Electron carrier of electron transport chain and free-radical-scavenging antioxidant | Slowdown in the decline of mitochondrial function in animal and cellular models of ALS | [163] |
| Mitochonic Acid 5 | Inductor of ATP synthase dimer formation | Increase in local ATP production and cell survival in various mitochondrial diseases | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Matveeva, L.A.; Belosludtsev, K.N. Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 16833. https://doi.org/10.3390/ijms242316833
Belosludtseva NV, Matveeva LA, Belosludtsev KN. Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2023; 24(23):16833. https://doi.org/10.3390/ijms242316833
Chicago/Turabian StyleBelosludtseva, Natalia V., Lyudmila A. Matveeva, and Konstantin N. Belosludtsev. 2023. "Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 24, no. 23: 16833. https://doi.org/10.3390/ijms242316833
APA StyleBelosludtseva, N. V., Matveeva, L. A., & Belosludtsev, K. N. (2023). Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 24(23), 16833. https://doi.org/10.3390/ijms242316833