
Ceramides Mediate Insulin-Induced Impairments in Cerebral Mitochondrial Bioenergetics in ApoE4 Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
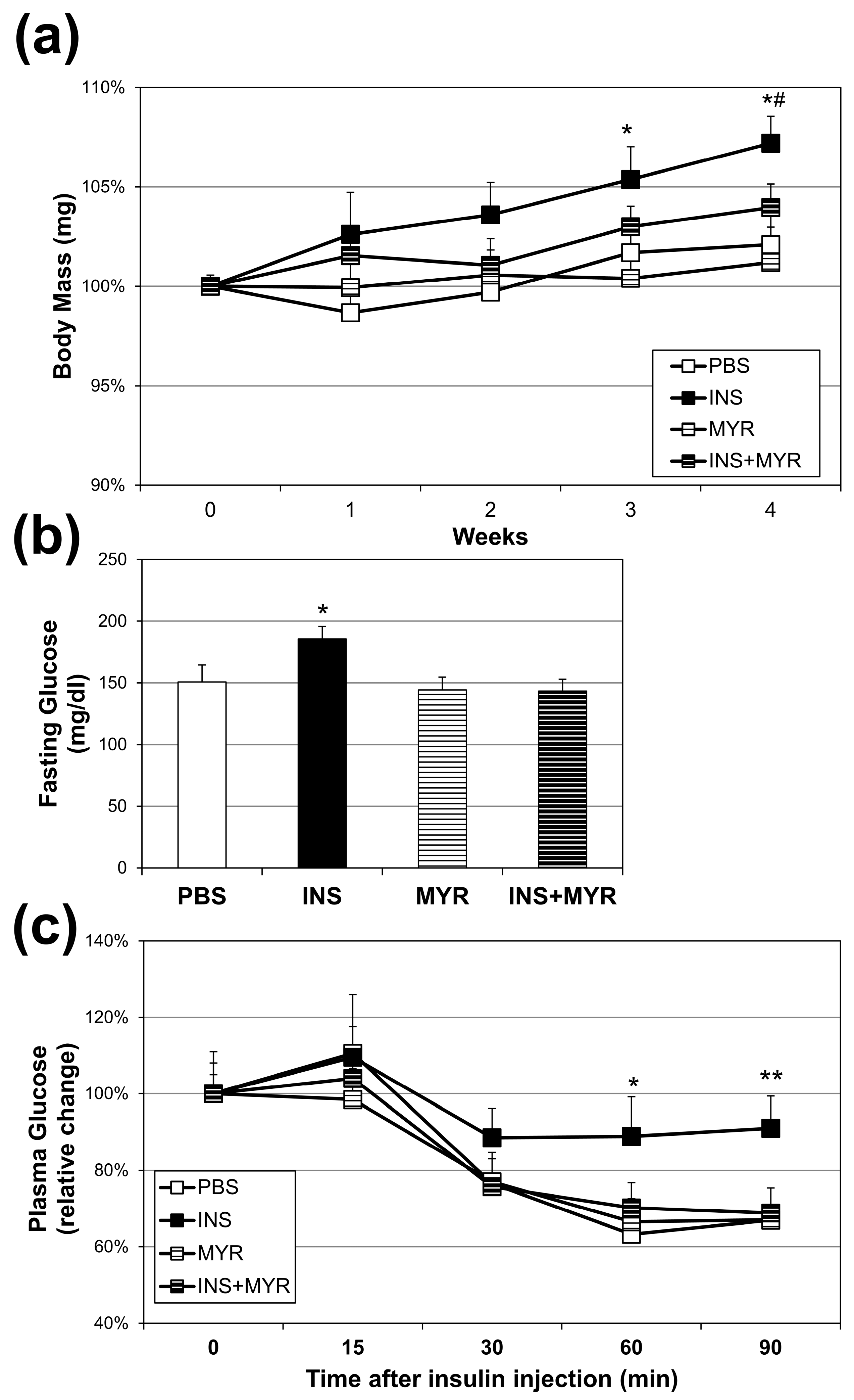
2.1. Chronic Insulin Injections Increase Body Weight and Reduce Insulin Tolerance in Male and Female ApoE4 Mice
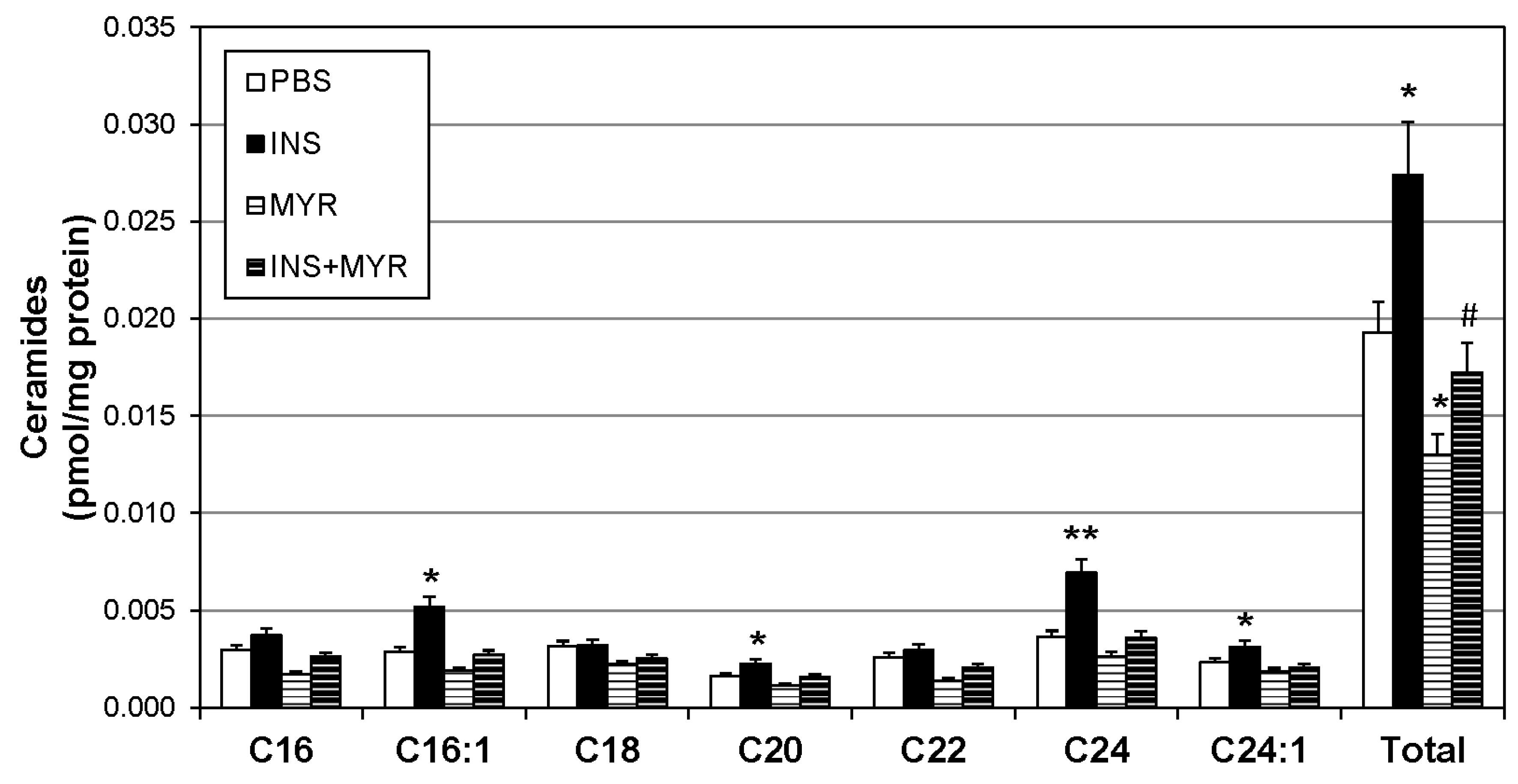
2.2. Insulin Increases Brain Ceramide Accrual in Male and Female ApoE4 Mice
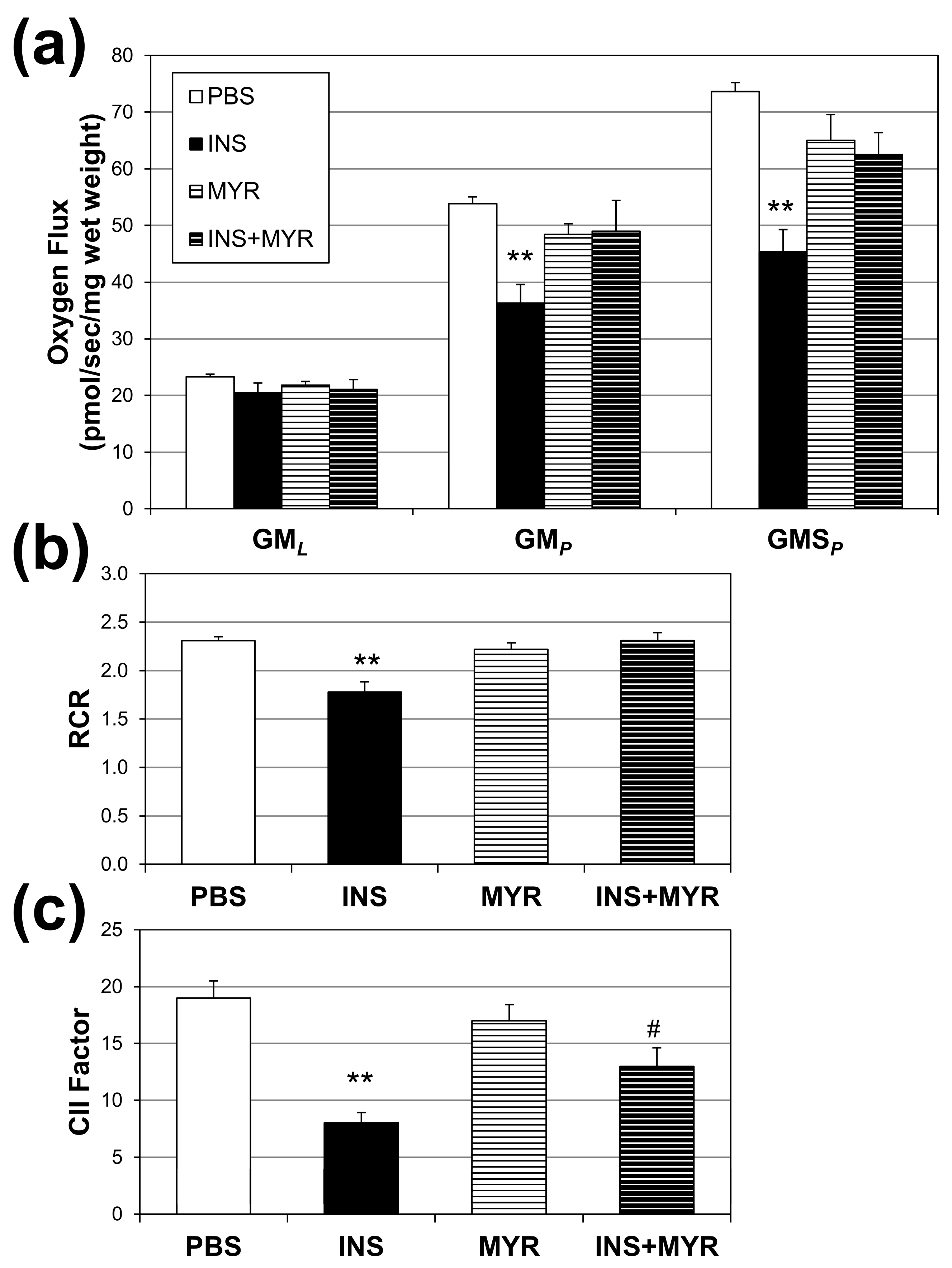
2.3. Insulin Disrupts Mitochondrial Function in Brain Tissue of Male and Female ApoE4 Mice
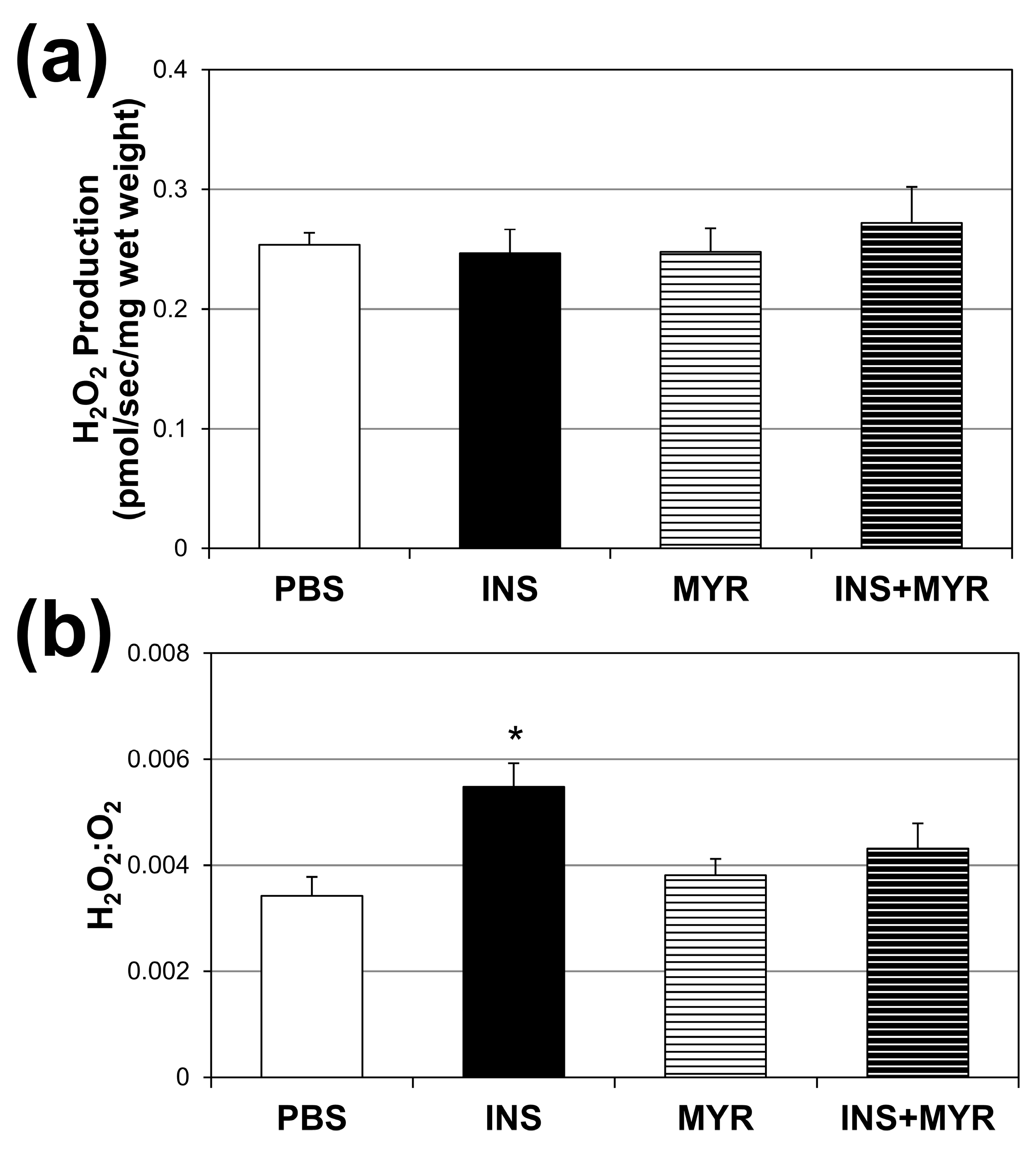
2.4. Chronic Insulin Injections Direct Brain O2 Use towards H2O2 Production in Male and Female ApoE4 Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Insulin Tolerance Test
4.3. Lipid Analysis
4.4. Mitochondrial Respirometry
4.5. H2O2 Emissions
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [PubMed]
- Katz, S.L.; MacLean, J.E.; Hoey, L.; Horwood, L.; Barrowman, N.; Foster, B.; Hadjiyannakis, S.; Legault, L.; Bendiak, G.N.; Kirk, V.G.; et al. Insulin Resistance and Hypertension in Obese Youth with Sleep-Disordered Breathing Treated with Positive Airway Pressure: A Prospective Multicenter Study. J. Clin. Sleep Med. 2017, 13, 1039–1047. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef]
- Crone, C. Facilitated transfer of glucose from blood into brain tissue. J. Physiol. 1965, 181, 103. [Google Scholar] [CrossRef]
- Hom, F.G.; Goodner, C.J.; Berrie, M.A. A (3H) 2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo: Validation of the model and the absence of an insulin effect on brain. Diabetes 1984, 33, 141–152. [Google Scholar] [CrossRef]
- Gerozissis, K. Brain insulin: Regulation, mechanisms of action and functions. Cell. Mol. Neurobiol. 2003, 23, 1–25. [Google Scholar] [CrossRef]
- Mueckler, M. Facilitative glucose transporters. Eur. J. Biochem. 1994, 219, 713–725. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Hopkins, D.; Williams, G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet. Med. 1997, 14, 1044–1050. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Chen, C.-M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 18, 700–789. [CrossRef]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef]
- Kuusisto, J.; Koivisto, K.; Mykkanen, L.; Helkala, E.L.; Vanhanen, M.; Hanninen, T.; Kervinen, K.; Kesaniemi, Y.A.; Riekkinen, P.J.; Laakso, M. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: Cross sectional population based study. BMJ 1997, 315, 1045–1049. [Google Scholar] [CrossRef]
- Sampath, D.; Sathyanesan, M.; Newton, S.S. Cognitive dysfunction in major depression and Alzheimer’s disease is associated with hippocampal-prefrontal cortex dysconnectivity. Neuropsychiatr. Dis. Treat. 2017, 13, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Blachnio-Zabielska, A. The role of ceramides in insulin resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Suzanne, M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475. [Google Scholar]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimer’s Dis. JAD 2012, 29, 537–547. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s disease: Key mediators of neuronal apoptosis induced by oxidative stress and Aβ accumulation. Oxidative Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Shubhra Chakrabarti, S.; Bir, A.; Poddar, J.; Sinha, M.; Ganguly, A.; Chakrabarti, S. Ceramide and sphingosine-1-phosphate in cell death pathways: Relevance to the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1232–1248. [Google Scholar] [CrossRef] [PubMed]
- Lyn-Cook, L.E., Jr.; Lawton, M.; Tong, M.; Silbermann, E.; Longato, L.; Jiao, P.; Mark, P.; Wands, J.R.; Xu, H.; de la Monte, S.M. Hepatic Ceramide May Mediate Brain Insulin Resistance and Neurodegeneration in Type 2 Diabetes and Non-alcoholic Steatohepatitis. J. Alzheimer’s Dis. 2009, 16, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.; Tippetts, T.S.; Anderson, M.C.; Holub, Z.E.; Moulton, E.R.; Swensen, A.C.; Prince, J.T.; Bikman, B.T. Insulin increases ceramide synthesis in skeletal muscle. J. Diabetes Res. 2014, 2014, 765784. [Google Scholar] [CrossRef]
- Hodson, A.E.; Tippetts, T.S.; Bikman, B.T. Insulin treatment increases myocardial ceramide accumulation and disrupts cardiometabolic function. Cardiovasc. Diabetol. 2015, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.-X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Sienski, G.A.-O.; Narayan, P.A.-O.; Bonner, J.A.-O.; Kory, N.A.-O.; Boland, S.A.-O.; Arczewska, A.A.; Ralvenius, W.A.-O.; Akay, L.A.-O.; Lockshin, E.A.-O.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13, eaaz4564. [Google Scholar] [CrossRef]
- Chaurasia, B.; Summers, S.A. Ceramides–lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef]
- Pontiroli, A.E.; Alberetto, M.; Pozza, G. Patients with insulinoma show insulin resistance in the absence of arterial hypertension. Diabetologia 1992, 35, 294–295. [Google Scholar] [CrossRef]
- Henry, R.R.; Gumbiner, B.; Ditzler, T.; Wallace, P.; Lyon, R.; Glauber, H.S. Intensive Conventional Insulin Therapy for Type II Diabetes: Metabolic effects during a 6-mo outpatient trial. Diabetes Care 1993, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S.; Leonetti, F.; Simonson, D.C.; Sheehan, P.; Matsuda, M.; DeFronzo, R.A. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia 1994, 37, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nevado-Holgado, A.; Whiley, L.; Snowden, S.G.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; Thambisetty, M. Association between plasma ceramides and phosphatidylcholines and hippocampal brain volume in late onset Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 60, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Rabins, P.V.; Lyketsos, C.G.; Carlson, M.C. Serum sphingomyelins and ceramides are early predictors of memory impairment. Neurobiol. Aging 2010, 31, 17–24. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Schech, S.; Carrick, R.; Carlson, M.C.; Mori, S.; Miller, M.I.; Ceritoglu, C.; Brown, T. Plasma ceramides are altered in mild cognitive impairment and predict cognitive decline and hippocampal volume loss. Alzheimer’s Dement. 2010, 6, 378–385. [Google Scholar] [CrossRef]
- McGrath, E.R.; Himali, J.J.; Xanthakis, V.; Duncan, M.S.; Schaffer, J.E.; Ory, D.S.; Peterson, L.R.; DeCarli, C.; Pase, M.P.; Satizabal, C.L. Circulating ceramide ratios and risk of vascular brain aging and dementia. Ann. Clin. Transl. Neurol. 2020, 7, 160–168. [Google Scholar] [CrossRef]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef]
- Kurz, J.; Parnham, M.J.; Geisslinger, G.; Schiffmann, S. Ceramides as novel disease biomarkers. Trends Mol. Med. 2019, 25, 20–32. [Google Scholar] [CrossRef]
- Teitsdottir, U.D.; Halldorsson, S.; Rolfsson, O.; Lund, S.H.; Jonsdottir, M.K.; Snaedal, J.; Petersen, P.H. Cerebrospinal fluid C18 ceramide associates with markers of Alzheimer’s disease and inflammation at the pre-and early stages of dementia. J. Alzheimer’s Dis. 2021, 81, 231–244. [Google Scholar] [CrossRef]
- Castellano, C.A.; Baillargeon, J.P.; Nugent, S.; Tremblay, S.; Fortier, M.; Imbeault, H.; Duval, J.; Cunnane, S.C. Regional Brain Glucose Hypometabolism in Young Women with Polycystic Ovary Syndrome: Possible Link to Mild Insulin Resistance. PLoS ONE 2015, 10, e0144116. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, E.; Hurtado-Carneiro, V.; LeBaut-Ayuso, Y.; Velazquez, E.; Garcia-Garcia, L.; Gomez-Oliver, F.; Ruiz-Albusac, J.M.; Avila, J.; Pozo, M.A. Significance of Brain Glucose Hypometabolism, Altered Insulin Signal Transduction, and Insulin Resistance in Several Neurological Diseases. Front. Endocrinol. 2022, 13, 873301. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, J.W.; Donato, L.J.; Kopecky, S.L.; Vasile, V.C.; Jaffe, A.S.; Laaksonen, R. Ceramides improve atherosclerotic cardiovascular disease risk assessment beyond standard risk factors. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 511, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Guan, Y.G.; Shui, G.H.; Siddique, M.M.; Holland, W.L.; Kim, J.Y.; Fabrias, G.; Wenk, M.R.; Summers, S.A. Fenretinide Prevents Lipid-induced Insulin Resistance by Blocking Ceramide Biosynthesis. J. Biol. Chem. 2012, 287, 17426–17437. [Google Scholar] [CrossRef]
- Li, Y.; Talbot, C.L.; Chandravanshi, B.; Ksiazek, A.; Sood, A.; Chowdhury, K.H.; Maschek, J.A.; Cox, J.; Babu, A.K.S.; Paz, H.A.; et al. Cordyceps inhibits ceramide biosynthesis and improves insulin resistance and hepatic steatosis. Sci. Rep. 2022, 12, 7273. [Google Scholar] [CrossRef]
- Erickson, K.A.; Smith, M.E.; Anthonymuthu, T.S.; Evanson, M.J.; Brassfield, E.S.; Hodson, A.E.; Bressler, M.A.; Tucker, B.J.; Thatcher, M.O.; Prince, J.T.; et al. AICAR inhibits ceramide biosynthesis in skeletal muscle. Diabetol. Metab. Syndr. 2012, 4, 45. [Google Scholar] [CrossRef]
- Ellison-Barnes, A.; Johnson, S.; Gudzune, K. Trends in obesity prevalence among adults aged 18 through 25 years, 1976–2018. JAMA 2021, 326, 2073–2074. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. In Mitochondrial Bioenergetics: Methods and Protocols; Palmeira, C.M., Moreno, A.J., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 25–58. [Google Scholar]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Napa, K.; Baeder, A.C.; Witt, J.E.; Rayburn, S.T.; Miller, M.G.; Dallon, B.W.; Gibbs, J.L.; Wilcox, S.H.; Winden, D.R.; Smith, J.H.; et al. LPS from P. gingivalis Negatively Alters Gingival Cell Mitochondrial Bioenergetics. Int. J. Dent. 2017, 2017, 2697210. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carr, S.T.; Saito, E.R.; Walton, C.M.; Saito, J.Y.; Hanegan, C.M.; Warren, C.E.; Trumbull, A.M.; Bikman, B.T. Ceramides Mediate Insulin-Induced Impairments in Cerebral Mitochondrial Bioenergetics in ApoE4 Mice. Int. J. Mol. Sci. 2023, 24, 16635. https://doi.org/10.3390/ijms242316635
Carr ST, Saito ER, Walton CM, Saito JY, Hanegan CM, Warren CE, Trumbull AM, Bikman BT. Ceramides Mediate Insulin-Induced Impairments in Cerebral Mitochondrial Bioenergetics in ApoE4 Mice. International Journal of Molecular Sciences. 2023; 24(23):16635. https://doi.org/10.3390/ijms242316635
Chicago/Turabian StyleCarr, Sheryl T., Erin R. Saito, Chase M. Walton, Jeremy Y. Saito, Cameron M. Hanegan, Cali E. Warren, Annie M. Trumbull, and Benjamin T. Bikman. 2023. "Ceramides Mediate Insulin-Induced Impairments in Cerebral Mitochondrial Bioenergetics in ApoE4 Mice" International Journal of Molecular Sciences 24, no. 23: 16635. https://doi.org/10.3390/ijms242316635
APA StyleCarr, S. T., Saito, E. R., Walton, C. M., Saito, J. Y., Hanegan, C. M., Warren, C. E., Trumbull, A. M., & Bikman, B. T. (2023). Ceramides Mediate Insulin-Induced Impairments in Cerebral Mitochondrial Bioenergetics in ApoE4 Mice. International Journal of Molecular Sciences, 24(23), 16635. https://doi.org/10.3390/ijms242316635