

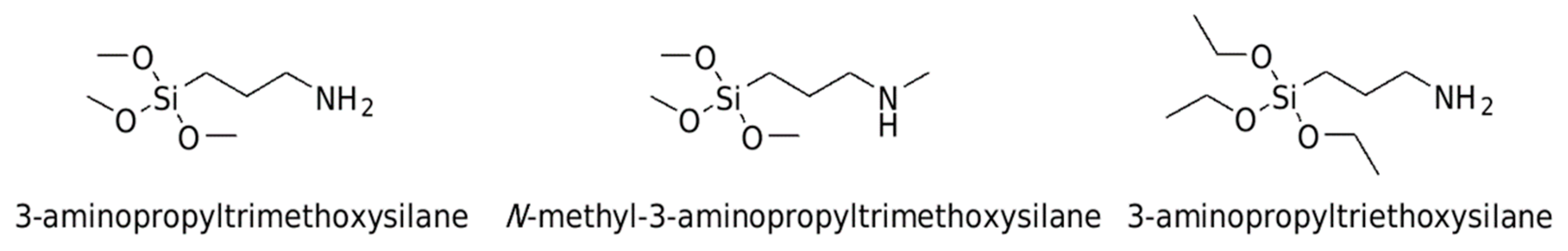
Intermolecular Interactions in 3-Aminopropyltrimethoxysilane, N-Methyl-3-aminopropyltrimethoxysilane and 3-Aminopropyltriethoxysilane: Insights from Computational Spectroscopy




Abstract
:1. Introduction
2. Results and Discussion
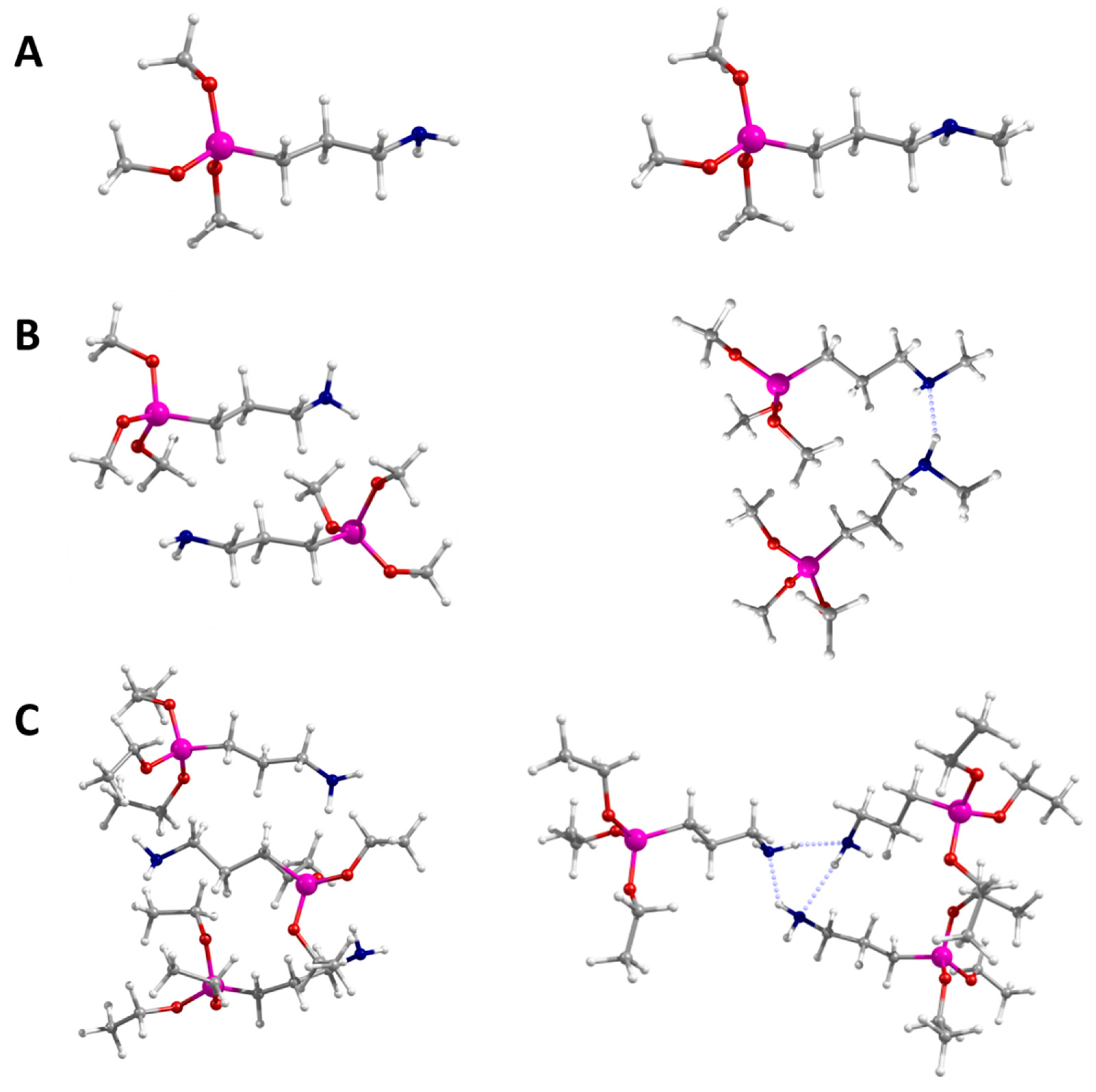
2.1. Calculated Structures: From Monomer to Trimer
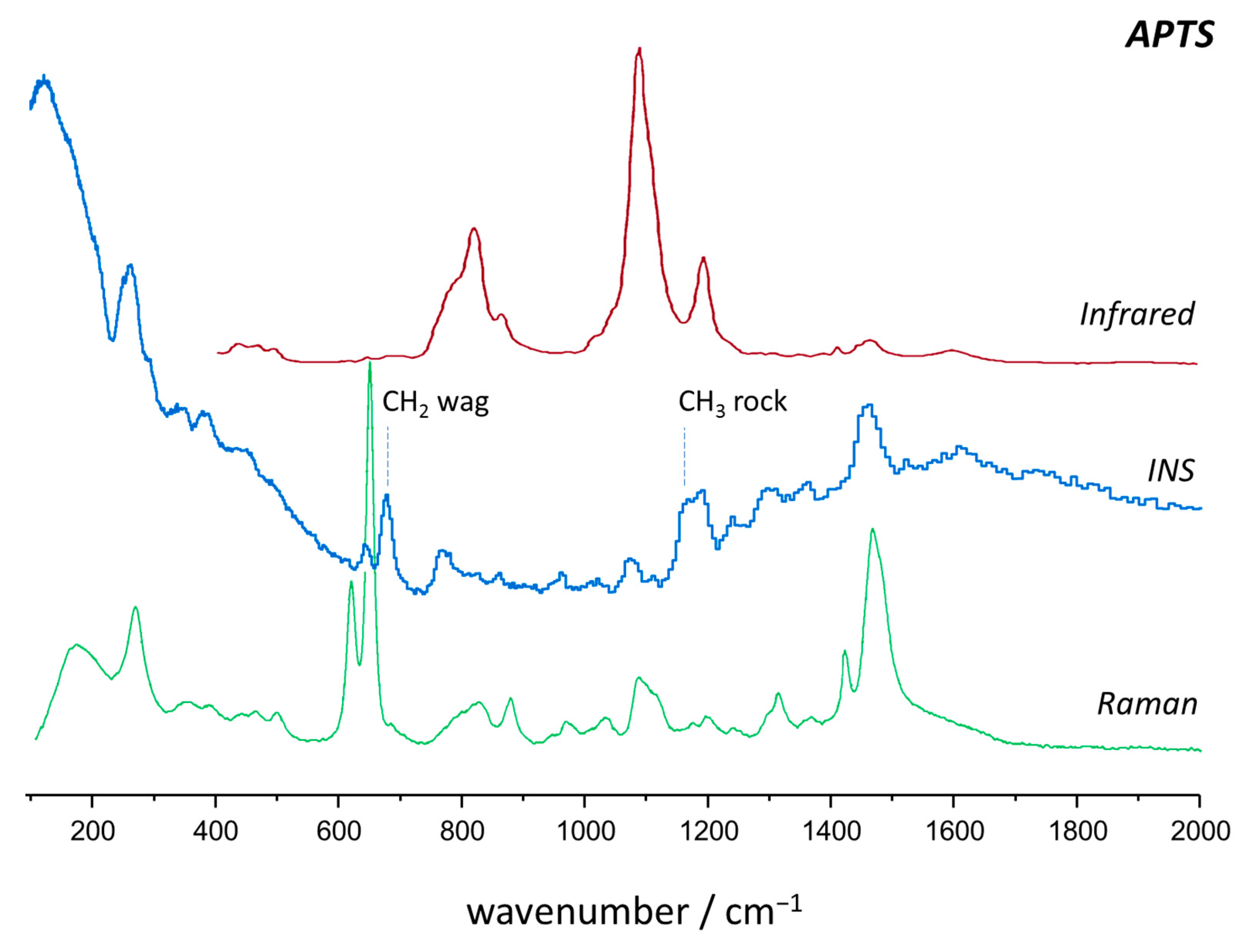
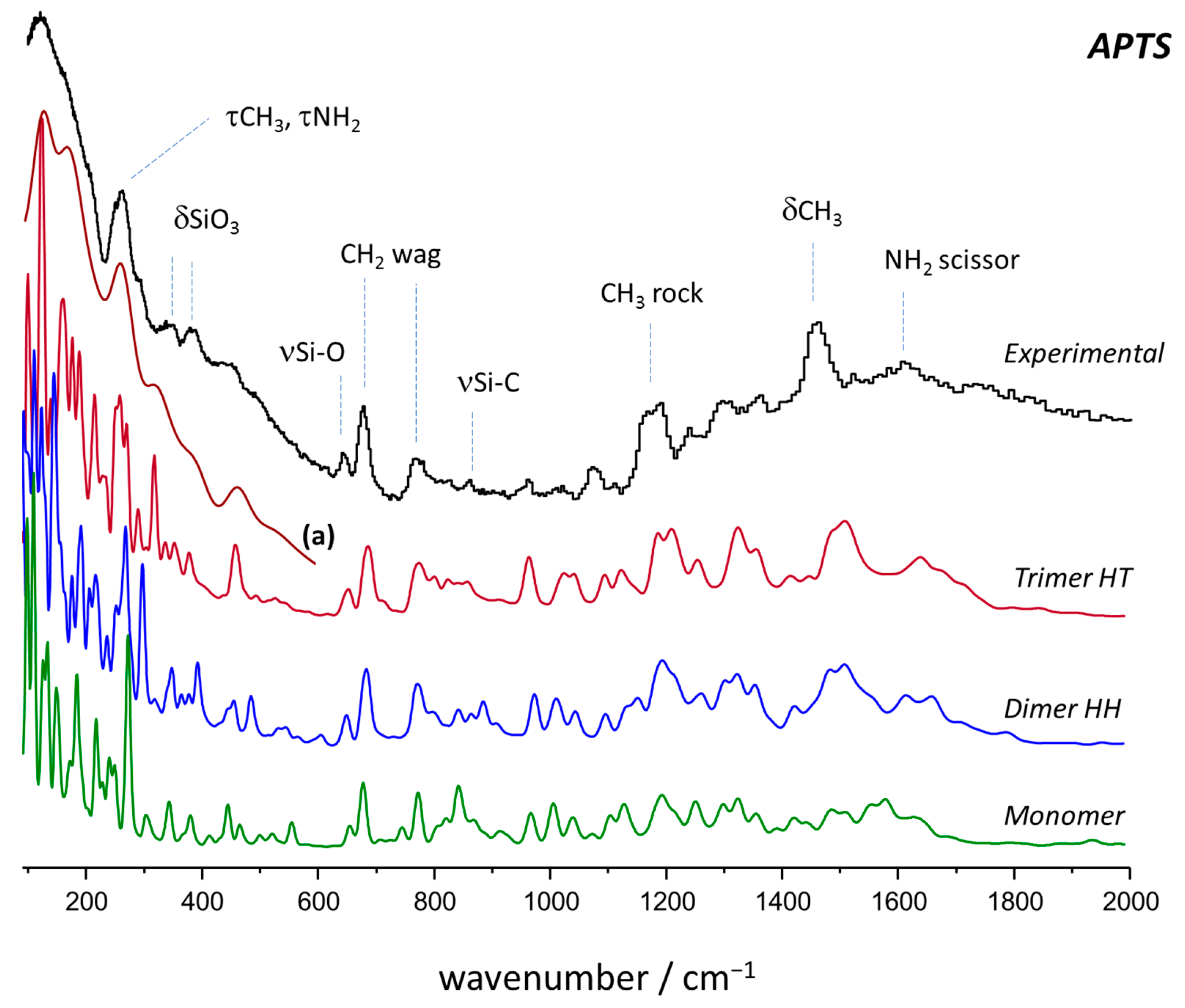
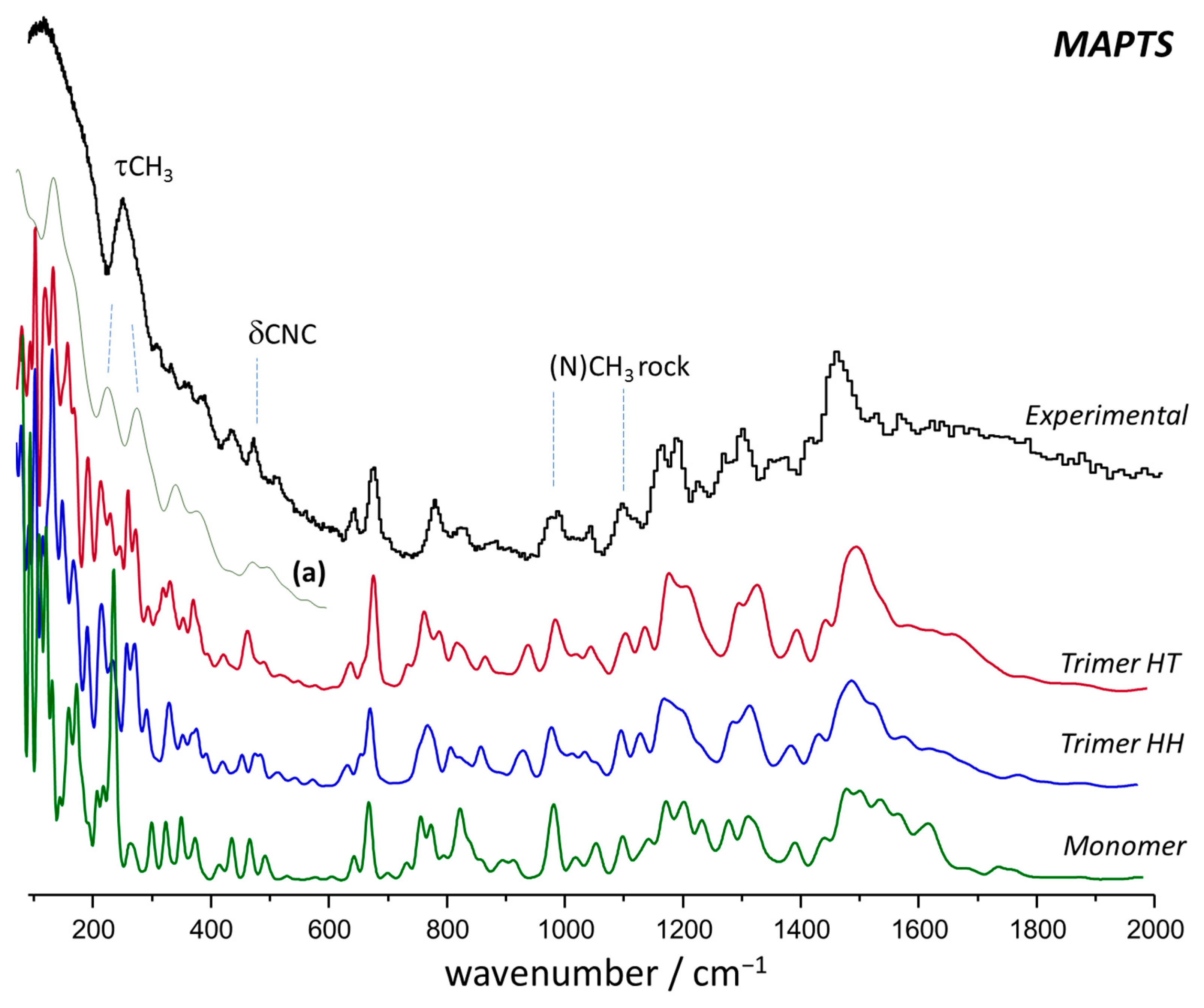
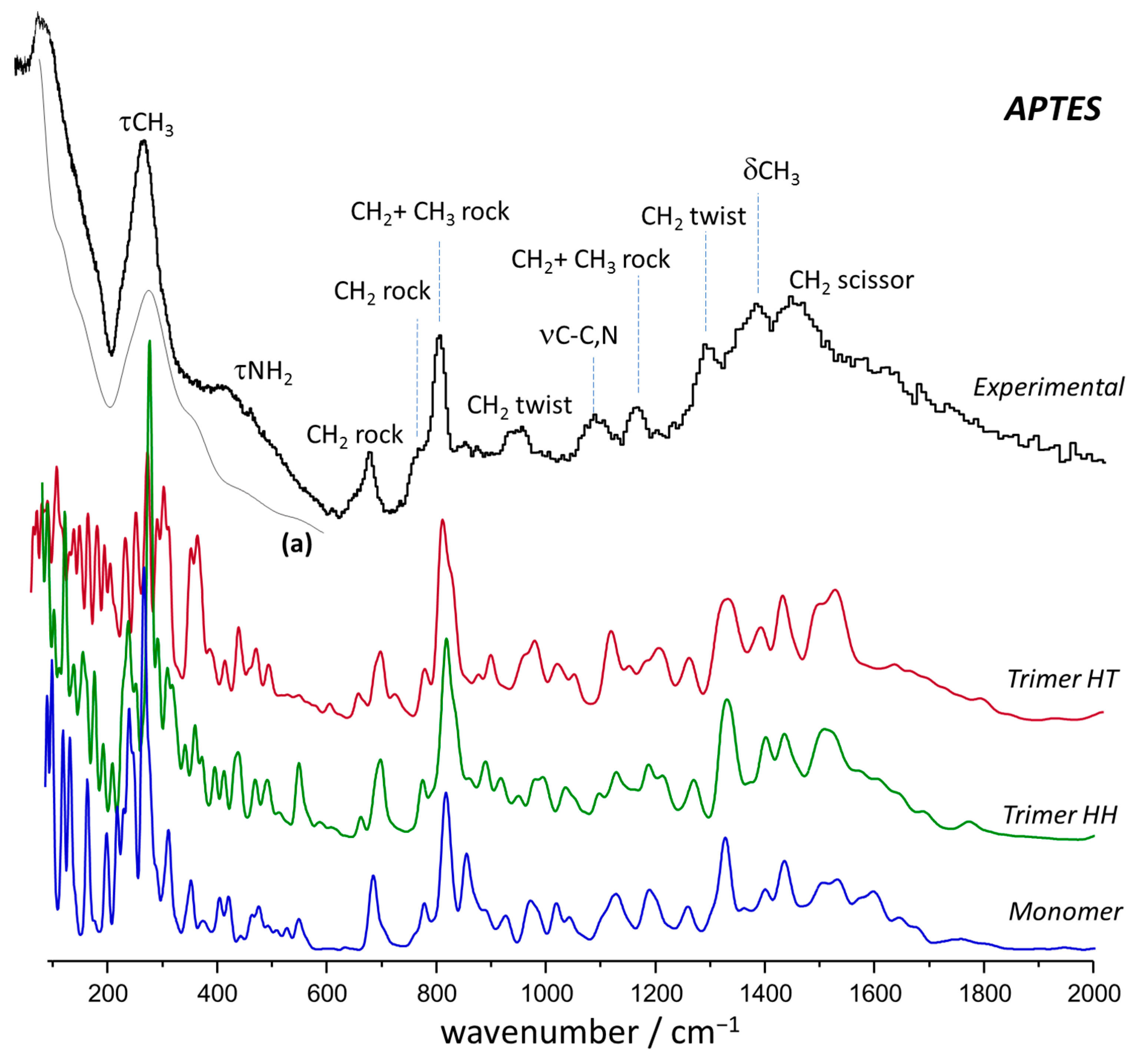
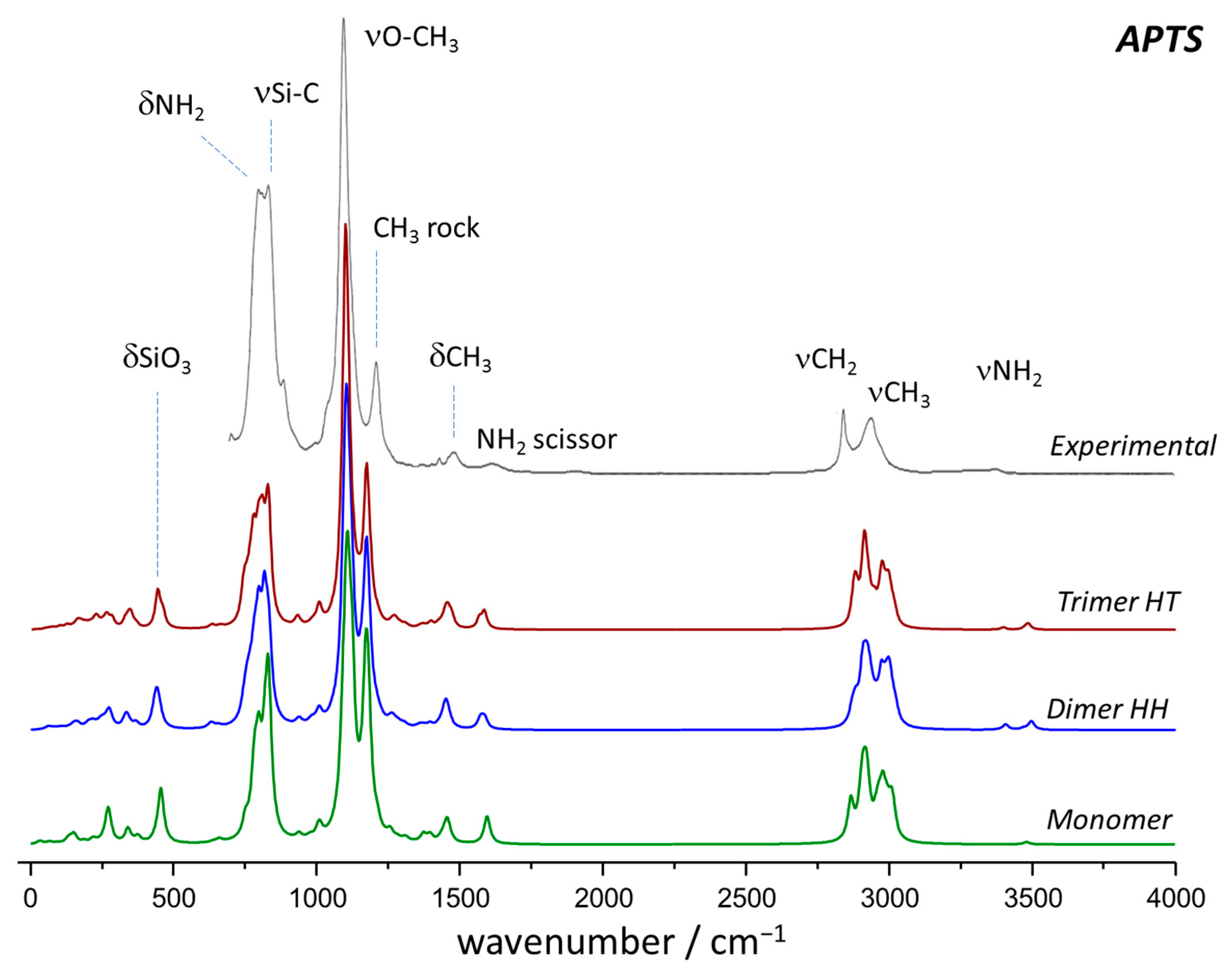
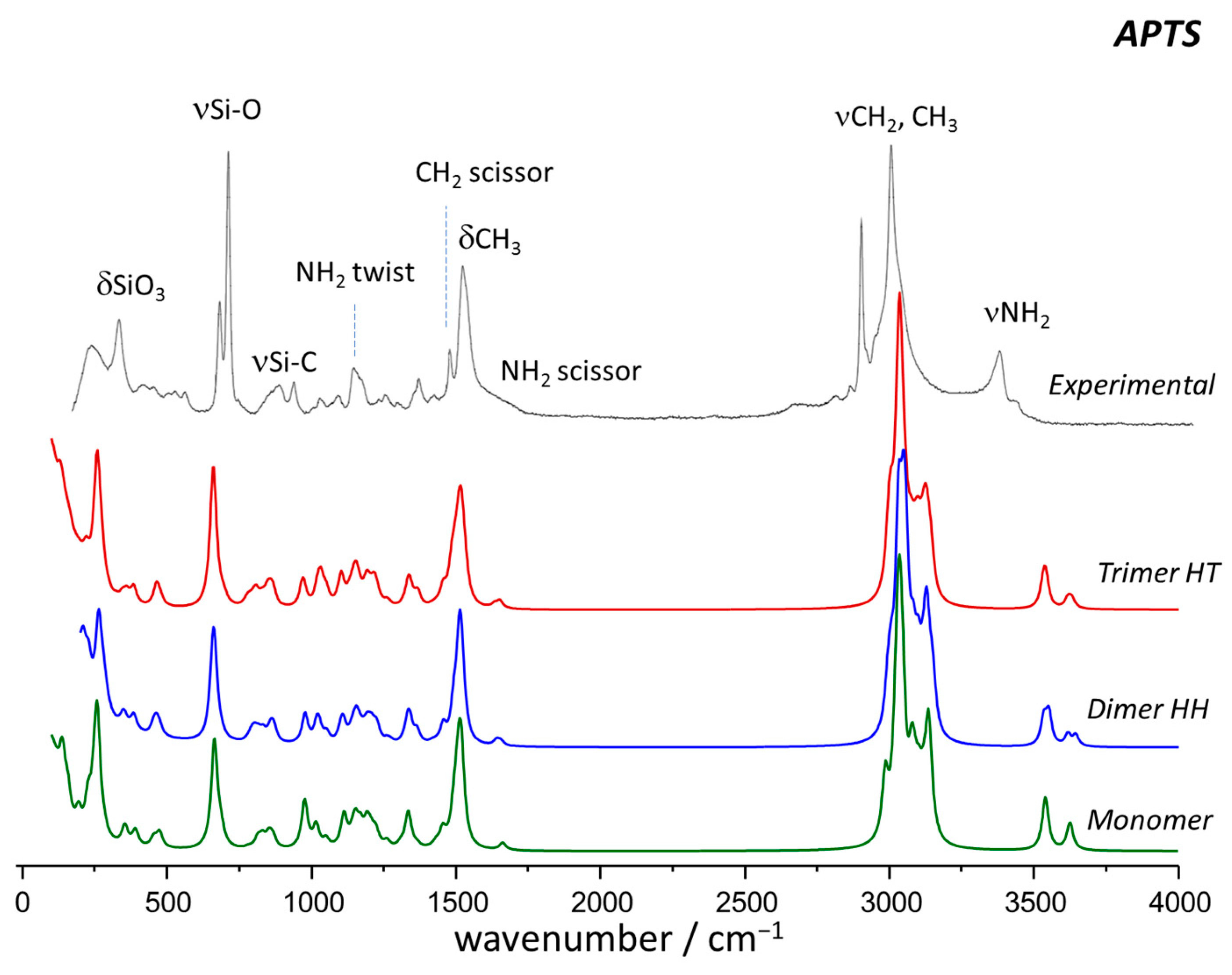
2.2. Vibrational Spectroscopy
2.3. Intensity Calculations: Photons vs. Neutrons
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gago, S.; Fernandes, J.A.; Rainho, J.P.; Sá Ferreira, R.A.; Pillinger, M.; Valente, A.A.; Santos, T.M.; Carlos, L.D.; Ribeiro-Claro, P.J.A.; Gonçalves, I.S. Highly luminescent tris(β-diketonate)europium(III) complexes immobilized in a functionalized mesoporous silica. Chem. Mater. 2005, 17, 5077–5084. [Google Scholar] [CrossRef]
- Ahangaran, F.; Navarchian, A.H. Recent advances in chemical surface modification of metal oxide nanoparticles with silane coupling agents: A review. Adv. Colloid Interface Sci. 2020, 286, 102298. [Google Scholar] [CrossRef]
- Khanjanzadeh, H.; Behrooz, R.; Bahramifar, N.; Gindl-Altmutter, W.; Bacher, M.; Edler, M.; Griesser, T. Surface chemical functionalization of cellulose nanocrystals by 3-aminopropyltriethoxysilane. Int. J. Biol. Macromol. 2018, 106, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Sim, Y.T.; Ruhaimi, A.H. Recent progress on (3-Aminopropyl)triethoxysilane (APTES) functionalized-adsorbent for CO2 capture. J. Phys. Conf. Ser. 2022, 2259, 12008. [Google Scholar] [CrossRef]
- Sypabekova, M.; Hagemann, A.; Rho, D.; Kim, S. Review: 3-Aminopropyltriethoxysilane (APTES) Deposition Methods on Oxide Surfaces in Solution and Vapor Phases for Biosensing Applications. Biosensors 2023, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.K.; Muehlberg, A.J.; Webster, D.C. Organic-inorganic hybrid coatings prepared from glycidyl carbamate resins and amino-functional silanes. Prog. Org. Coat. 2008, 63, 405–415. [Google Scholar] [CrossRef]
- Cai, J.; Liu, Y.; Shu, X. Long-Period Fiber Grating Sensors for Chemical and Biomedical Applications. Sensors 2023, 23, 542. [Google Scholar] [CrossRef]
- Kurczewska, J.; Ceglowski, M.; Messyasz, B.; Schroeder, G. Dendrimer-functionalized halloysite nanotubes for effective drug delivery. Appl. Clay Sci. 2018, 153, 134–143. [Google Scholar] [CrossRef]
- Wu, X.; Jia, H.; Fu, W.; Li, M.; Pan, Y. Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane-Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application. Molecules 2023, 28, 2464. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, E.P.C. Chemical Functionalities of 3-aminopropyltriethoxy-silane for Surface Modification of Metal Oxide Nanoparticles. Silicon 2022, 14, 6535–6545. [Google Scholar] [CrossRef]
- Jie, W.; Yan-Hua, C.; Jia, Z. Synthesis and Fluorescence Properties of Water-Solube ZnO/Aminopropyl-Siloxane Quantum Dots. Chin. J. Inorg. Chem. 2010, 26, 1199–1206. [Google Scholar]
- Giudice, C.A.; Alfieri, P.V.; Canosa, G. Siloxanes synthesized “in situ” by sol-gel process for fire control in wood of Araucaria angustifolia. Fire Saf. J. 2013, 61, 348–354. [Google Scholar] [CrossRef]
- Yu, M.; Lu, Q.; Cui, Z.; Wang, X.; Ge, F.; Wang, X. Siloxane-epoxy composite coatings for enhanced resistance to large temperature variations. Prog. Org. Coat. 2020, 139, 105457. [Google Scholar] [CrossRef]
- Maslennikova, V.V.; Filatov, S.N.; Orlov, A.V.; Surin, N.M.; Svidchenko, E.A.; Chistyakov, E.M. Luminescent Coatings Based on (3-Aminopropyl)triethoxysilane and Europium Complex β-Diketophosphazene. Polymers 2022, 14, 728. [Google Scholar] [CrossRef] [PubMed]
- Alsalbokh, M.; Fakeri, N.; Lawson, S.; Rownaghi, A.A.; Rezaei, F. Adsorption of iodine from aqueous solutions by aminosilane-grafted mesoporous alumina. Chem. Eng. J. 2021, 415, 128968. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, Y.R.; Jeong, W.Y.; Lee, S.; Shin, J.H.; Lee, G.-J. Potential Protective Effect of Nitric Oxide-Releasing Nanofibers in Hypoxia/Reoxygenation-Induced Cardiomyocyte Injury. J. Nanosci. Nanotechnol. 2019, 19, 6539–6545. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Stansbury, J.W. Photopolymerization shrinkage-stress reduction in polymer-based dental restoratives by surface modification of fillers. Dent. Mater. 2021, 37, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, W.K.; Chiang, K.-Y. Amine-functionalized biogenic silica incorporation effect on poly (ether-block-amide) membrane CO2/N2 separation performance. J. Memb. Sci. 2023, 680, 121732. [Google Scholar] [CrossRef]
- Frank, B.P.; Durkin, D.P.; Caudill, E.R.; Zhu, L.; White, D.H.; Curry, M.L.; Pedersen, J.A.; Fairbrother, D.H. Impact of Silanization on the Structure, Dispersion Properties, and Biodegradability of Nanocellulose as a Nanocomposite Filler. ACS Appl. Nano Mater. 2018, 1, 7025–7038. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, F.; Dai, J.; Zhang, Q.; Jiang, M.; Xiong, Y. Recycled-Oil-Based Polyurethane Modified with Organic Silicone for Controllable Release of Coated Fertilizer. Polymers 2019, 11, 454. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Zhao, X.; Fu, J.; Zhang, G.; Ma, W.; Zhang, W.-H.; Choi, H.W. Surface passivation of halide perovskite nanocrystals for stable and high purity color conversion. Appl. Phys. Lett. 2022, 120, 01190. [Google Scholar] [CrossRef]
- Jimenez-Ruiz, M.; Ivanov, A.; Fuard, S. LAGRANGE—The new neutron vibrational spectrometer at the ILL. J. Phys. Conf. Ser. 2014, 549, 012004. [Google Scholar] [CrossRef]
- Pinna, R.S.; Rudić, S.; Parker, S.F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S.P.; Zacek, D.; Smith, C.A.; Capstick, M.J.; et al. The neutron guide upgrade of the TOSCA spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2018, 896, 68–74. [Google Scholar] [CrossRef]
- Seeger, P.A.; Daemen, L.L.; Larese, J.Z. Resolution of VISION, a crystal-analyzer spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2009, 604, 719–728. [Google Scholar] [CrossRef]
- Harrelson, T.F.; Cheng, Y.Q.; Li, J.; Jacobs, I.E.; Ramirez-Cuesta, A.J.; Faller, R.; Moulé, A.J. Identifying atomic scale structure in undoped/doped semicrystalline P3HT using Inelastic Neutron Scattering. Macromolecules 2017, 50, 2424–2435. [Google Scholar] [CrossRef]
- Cavalcante, L.S.R.; Dettmann, M.A.; Sours, T.; Yang, D.; Daemen, L.L.; Gates, B.C.; Kulkarni, A.R.; Moule, A.J. Elucidating correlated defects in metal organic frameworks using theory-guided inelastic neutron scattering spectroscopy. Mater. Horiz. 2023, 10, 187–196. [Google Scholar] [CrossRef]
- Nolasco, M.M.; Rodrigues, L.C.; Araujo, C.F.; Coimbra, M.M.; Ribeiro-Claro, P.; Vaz, P.D.; Rudic, S.; Silvestre, A.J.D.; Bouyahya, C.; Majdoub, M.; et al. From PEF to PBF: What difference does the longer alkyl chain make a computational spectroscopy study of poly(butylene 2,5-furandicarboxylate). Front. Chem. 2022, 10, 1056286. [Google Scholar] [CrossRef]
- Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Vaz, P.D. Vibrational Dynamics in crystalline 4-(dimethylamino) benzaldehyde: Inelastic Neutron Scattering and Periodic DFT Study. Materials 2022, 15, 475. [Google Scholar] [CrossRef]
- Nolasco, M.M.; Coimbra, M.M.; Parker, S.F.; Vaz, P.D.; Ribeiro-Claro, P.J.A. Structural Dynamics of Chloromethanes through Computational Spectroscopy: Combining INS and DFT. Molecules 2022, 27, 7661. [Google Scholar] [CrossRef]
- Drużbicki, K.; Mikuli, E.; Pałka, N.; Zalewski, S.; Ossowska-Chruściel, M.D. Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Pawlukojć, A.; Hetmańczyk, Ł. INS, DFT and temperature dependent IR studies on dynamical properties of acetylcholine chloride. Vib. Spectrosc. 2016, 82, 37–43. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific Publishing Co.: Singapore, 2005; ISBN 978-981-256-013-1. [Google Scholar]
- Parker, S.F.; Lennon, D.; Albers, P.W. Vibrational Spectroscopy with Neutrons: A Review of New Directions. Appl. Spectrosc. 2011, 65, 1325–1341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Yamaguchi, Y.; Gaw, J.F.; Schaefer, H.F.; Binkley, J.S. Analytic raman intensities from molecular electronic wave-functions. J. Chem. Phys. 1986, 84, 531–532. [Google Scholar] [CrossRef]
- Porezag, D.; Pederson, M.R. Infrared intensities and Raman-scattering activities within density-functional theory. Phys. Rev. B 1996, 54, 7830–7836. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A.J.; Tomkinson, J.; Rudic, S.; Pinna, R.S.; Gorini, G.; Fernandez Castanon, J. Recent and future developments on TOSCA at ISIS. J. Phys. Conf. Ser. 2014, 554, 012003. [Google Scholar] [CrossRef]
- ISIS Facility INS/TOSCA. Available online: https://www.isis.stfc.ac.uk/Pages/tosca.aspx (accessed on 15 September 2022).
- Nolasco, M.M.; Pedro, S.N.; Vilela, C.; Vaz, P.D.; Ribeiro-Claro, P.; Rudic, S.; Parker, S.F.; Freire, C.S.R.; Freire, M.G.; Silvestre, A.J.D. Water in Deep Eutectic Solvents: New Insights from Inelastic Neutron Scattering Spectroscopy. Front. Phys. 2022, 10, 834571. [Google Scholar] [CrossRef]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.M.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Leal, R.F.; Gigg, M.A.; et al. Mantid-Data analysis and visualization package for neutron scattering and mu SR experiments. Nucl. Instrum. Methods Phys. Res. A 2014, 764, 156–166. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- NIST. Computational Chemistry Comparison and Benchmark Database. Available online: https://cccbdb.nist.gov/vibscalejustx.asp (accessed on 15 September 2022).
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The modern software for INS interpretation. Phys. B Condens. Matter 2018, 551, 443–448. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance/pm | APTS HH | APTS HT | MAPTS HH | MAPTS HT | APTES HH | APTES HT |
|---|---|---|---|---|---|---|
| Dimer | ||||||
| N–H···N | 225 | 225 | 224 | |||
| C–H···O | 264 | 261 | 263 | |||
| N–H···O | 223 | 225 | 221 | |||
| Trimer | ||||||
| N–H···N | 221 | 216 | 219 | |||
| C–H···O | 250 | 254 | 248 | |||
| N–H···O | 236 | 221 | 222 | |||
| C–H···N | 200 | 239 | 244 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolasco, M.M.; Parker, S.F.; Vaz, P.D.; Ribeiro-Claro, P.J.A. Intermolecular Interactions in 3-Aminopropyltrimethoxysilane, N-Methyl-3-aminopropyltrimethoxysilane and 3-Aminopropyltriethoxysilane: Insights from Computational Spectroscopy. Int. J. Mol. Sci. 2023, 24, 16634. https://doi.org/10.3390/ijms242316634
Nolasco MM, Parker SF, Vaz PD, Ribeiro-Claro PJA. Intermolecular Interactions in 3-Aminopropyltrimethoxysilane, N-Methyl-3-aminopropyltrimethoxysilane and 3-Aminopropyltriethoxysilane: Insights from Computational Spectroscopy. International Journal of Molecular Sciences. 2023; 24(23):16634. https://doi.org/10.3390/ijms242316634
Chicago/Turabian StyleNolasco, Mariela M., Stewart F. Parker, Pedro D. Vaz, and Paulo J. A. Ribeiro-Claro. 2023. "Intermolecular Interactions in 3-Aminopropyltrimethoxysilane, N-Methyl-3-aminopropyltrimethoxysilane and 3-Aminopropyltriethoxysilane: Insights from Computational Spectroscopy" International Journal of Molecular Sciences 24, no. 23: 16634. https://doi.org/10.3390/ijms242316634
APA StyleNolasco, M. M., Parker, S. F., Vaz, P. D., & Ribeiro-Claro, P. J. A. (2023). Intermolecular Interactions in 3-Aminopropyltrimethoxysilane, N-Methyl-3-aminopropyltrimethoxysilane and 3-Aminopropyltriethoxysilane: Insights from Computational Spectroscopy. International Journal of Molecular Sciences, 24(23), 16634. https://doi.org/10.3390/ijms242316634