

Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
2.1. Characteristics and Information of Patient-Derived PTC Cell Lines
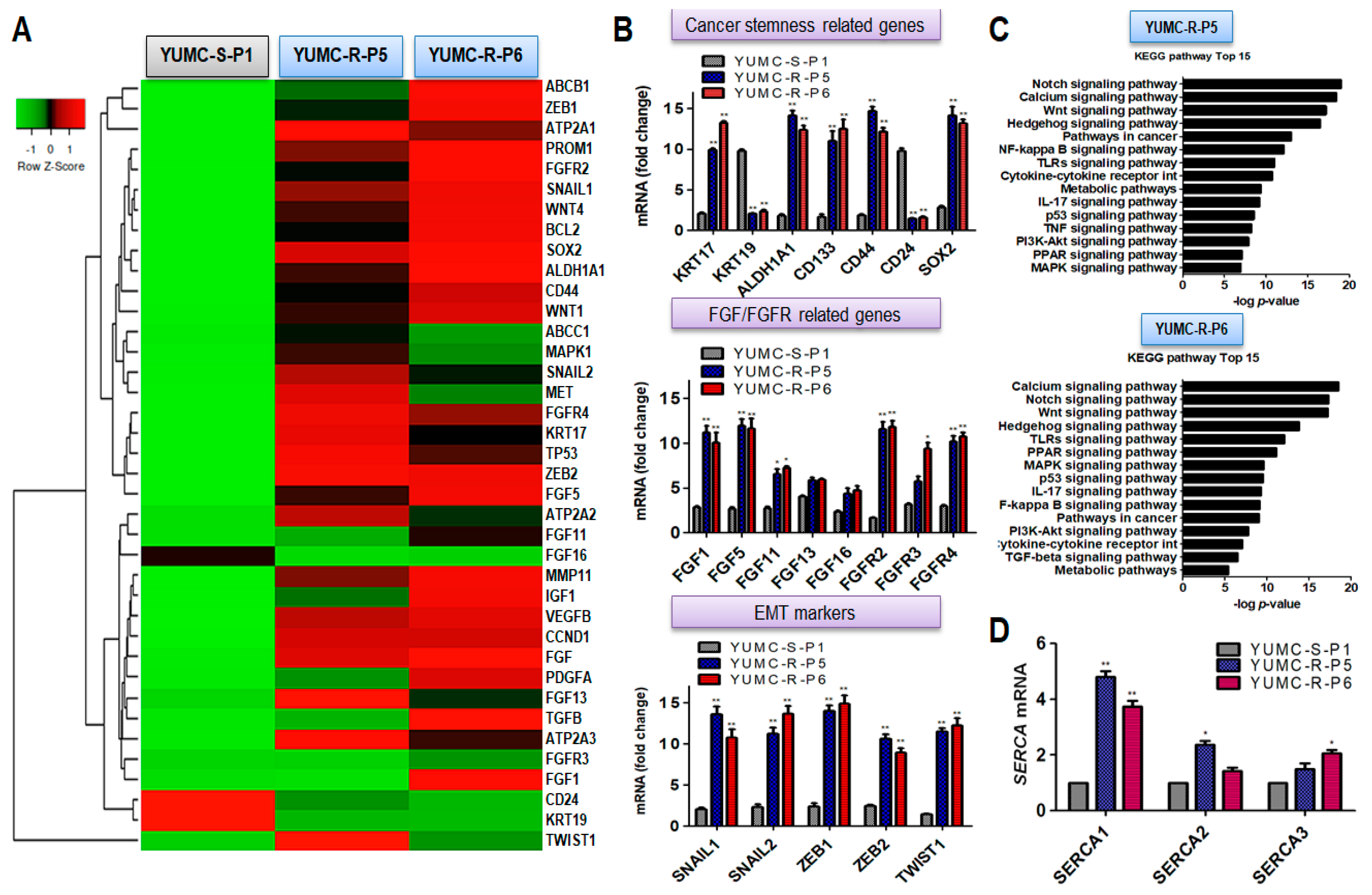
2.2. A Dissimilarity of Genetic Change and Stimulated Signaling Pathways between Patient-Derived Sorafenib-Sensitive and Sorafenib-Resistant PTC Cell Lines
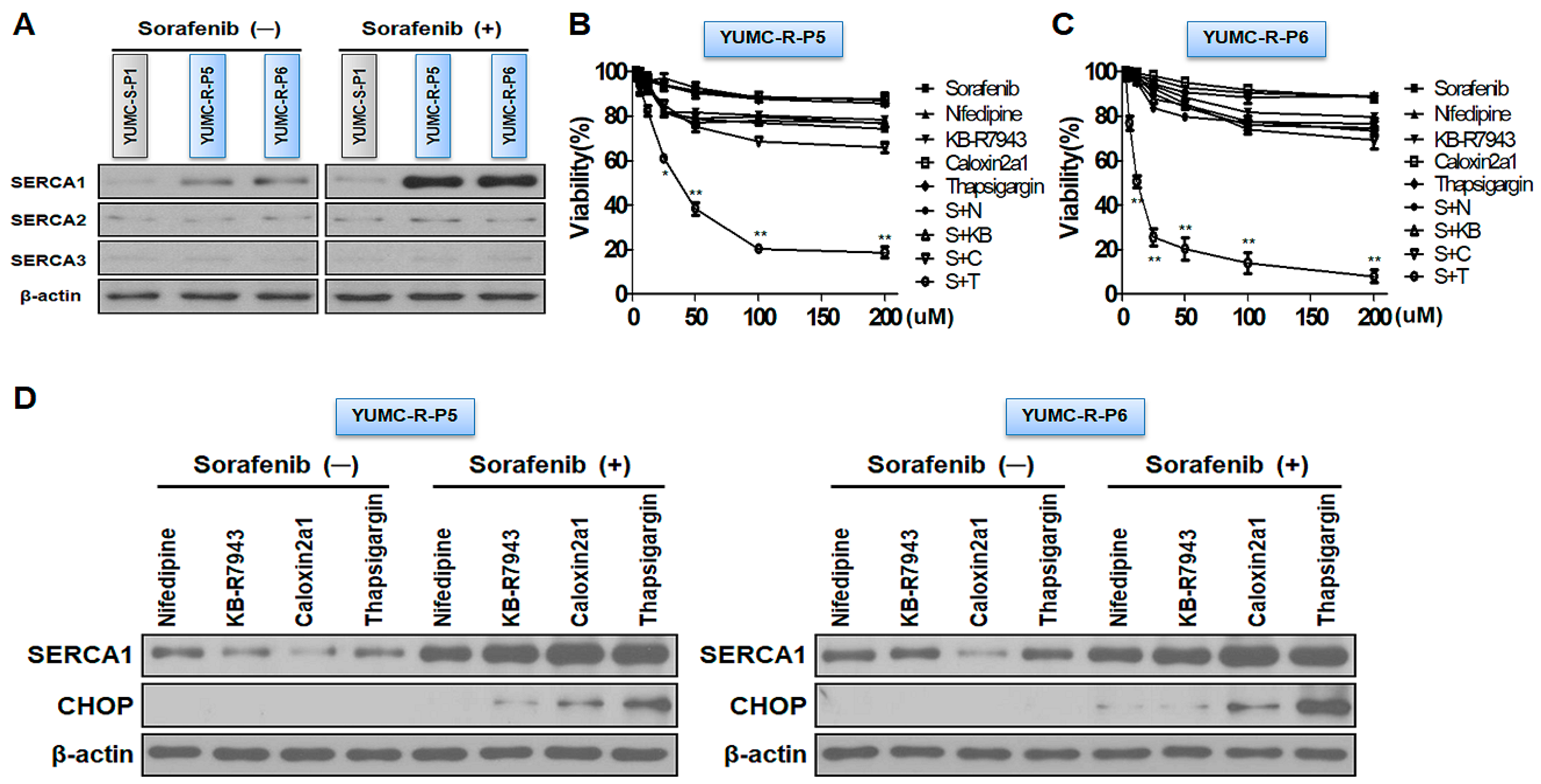
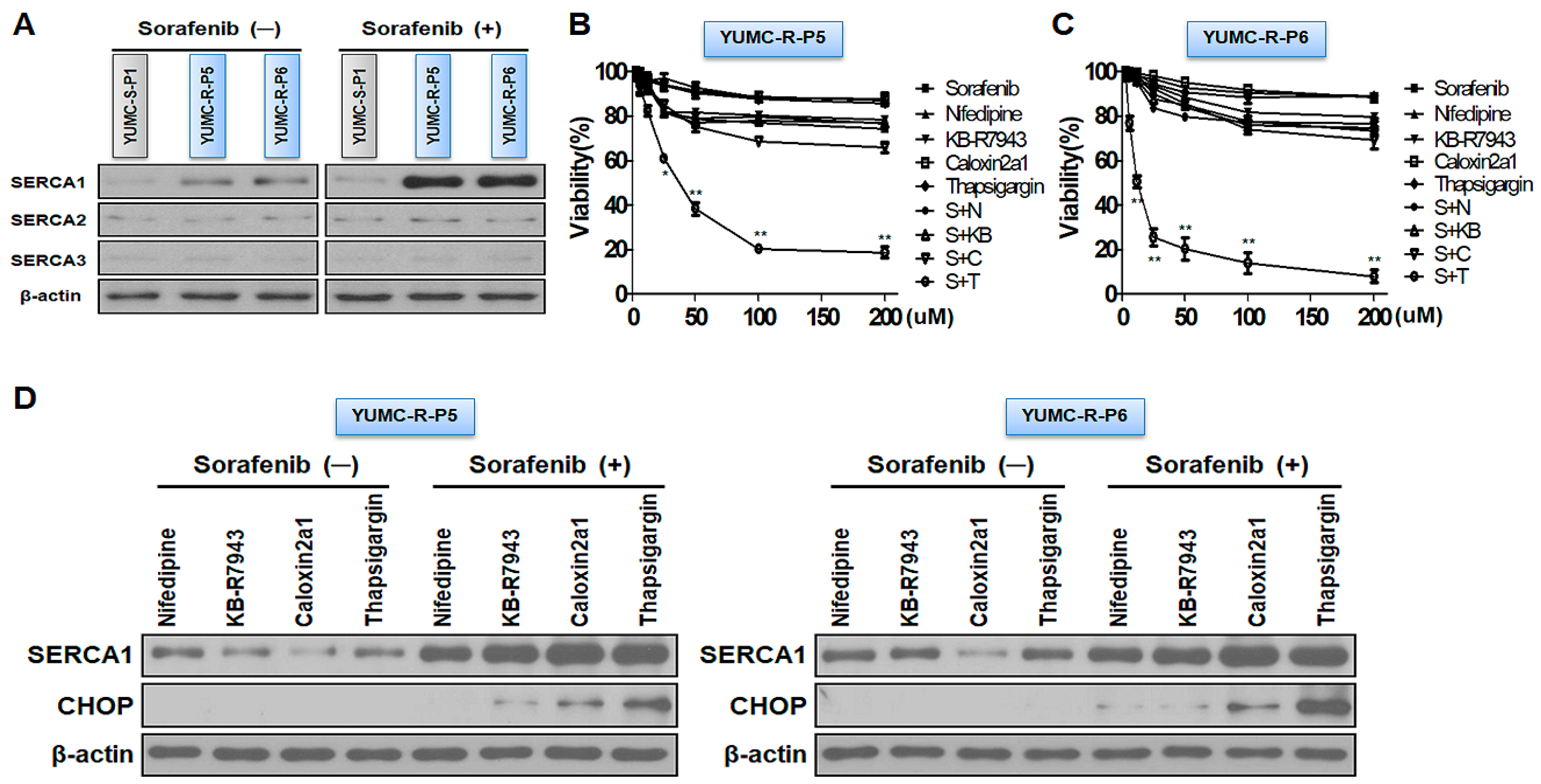
2.3. SERCAs Being Key Regulators to Prolong Survival under Sorafenib-Treated Conditions in Sorafenib-Resistant PTC Cells
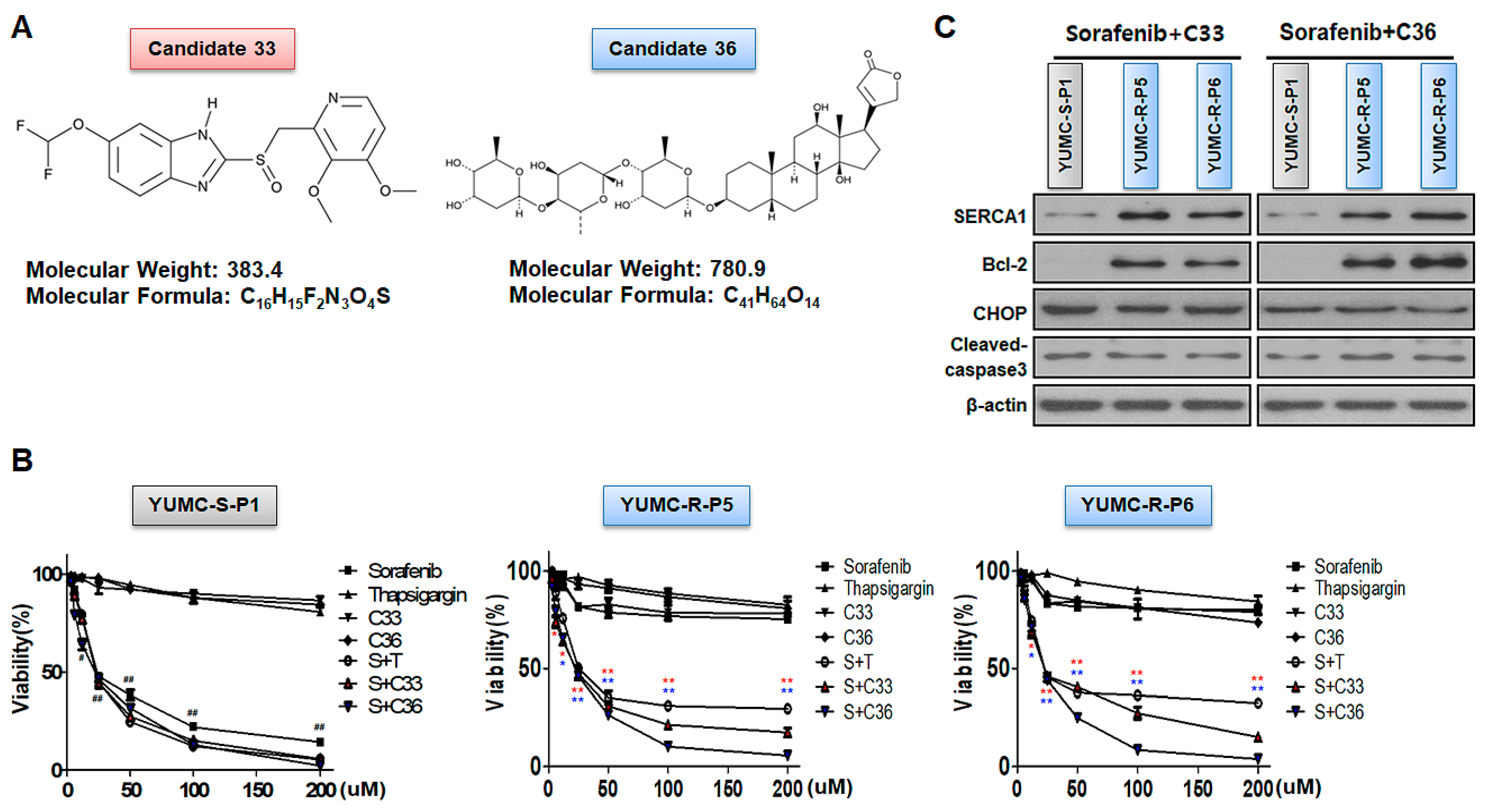
2.4. SERCA Target Specific Discovery of the New Therapeutic Access of Candidates 33 and 36 by In-Silico Screening for Suppression of Patient-Derived Sorafenib-Resistant PTC
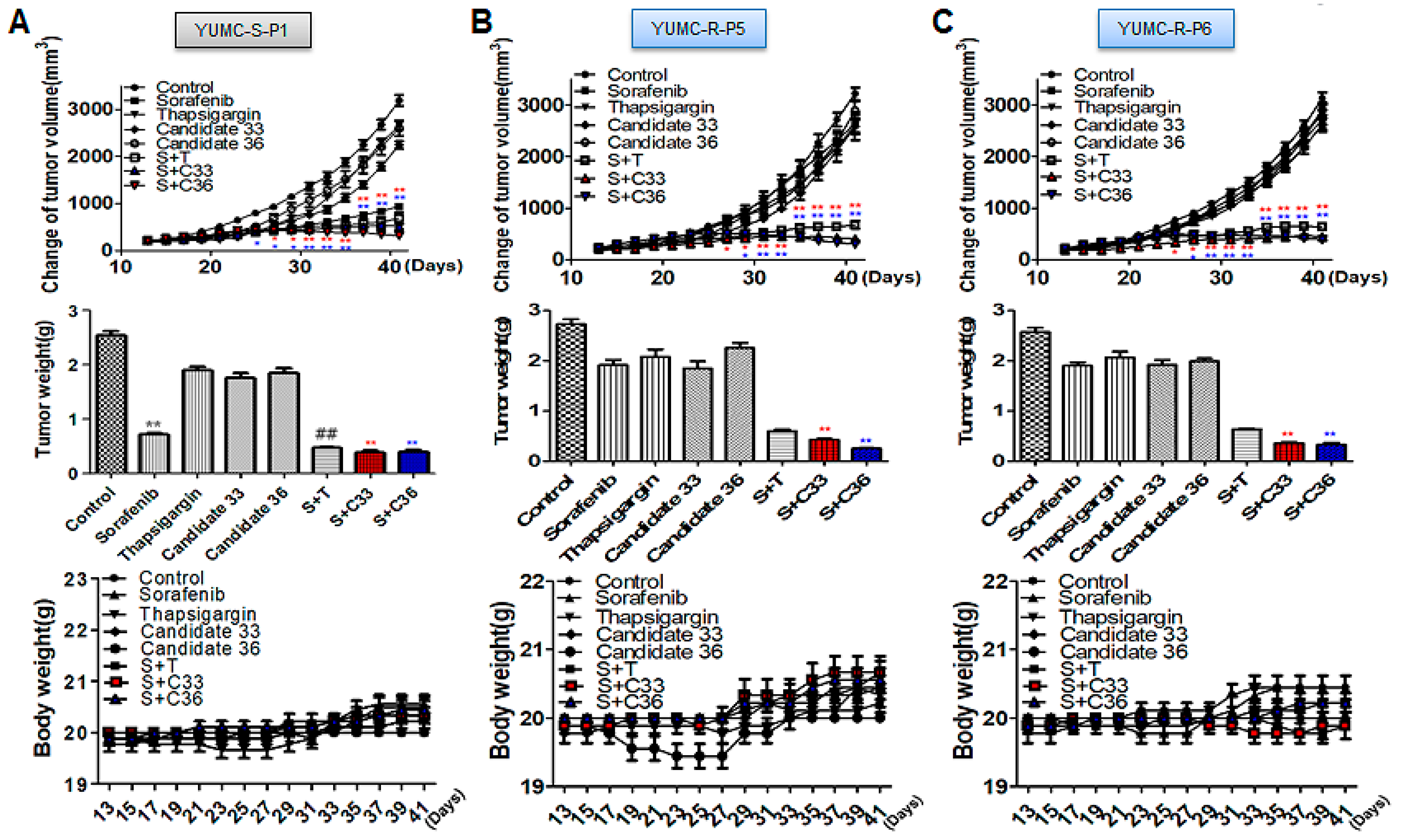
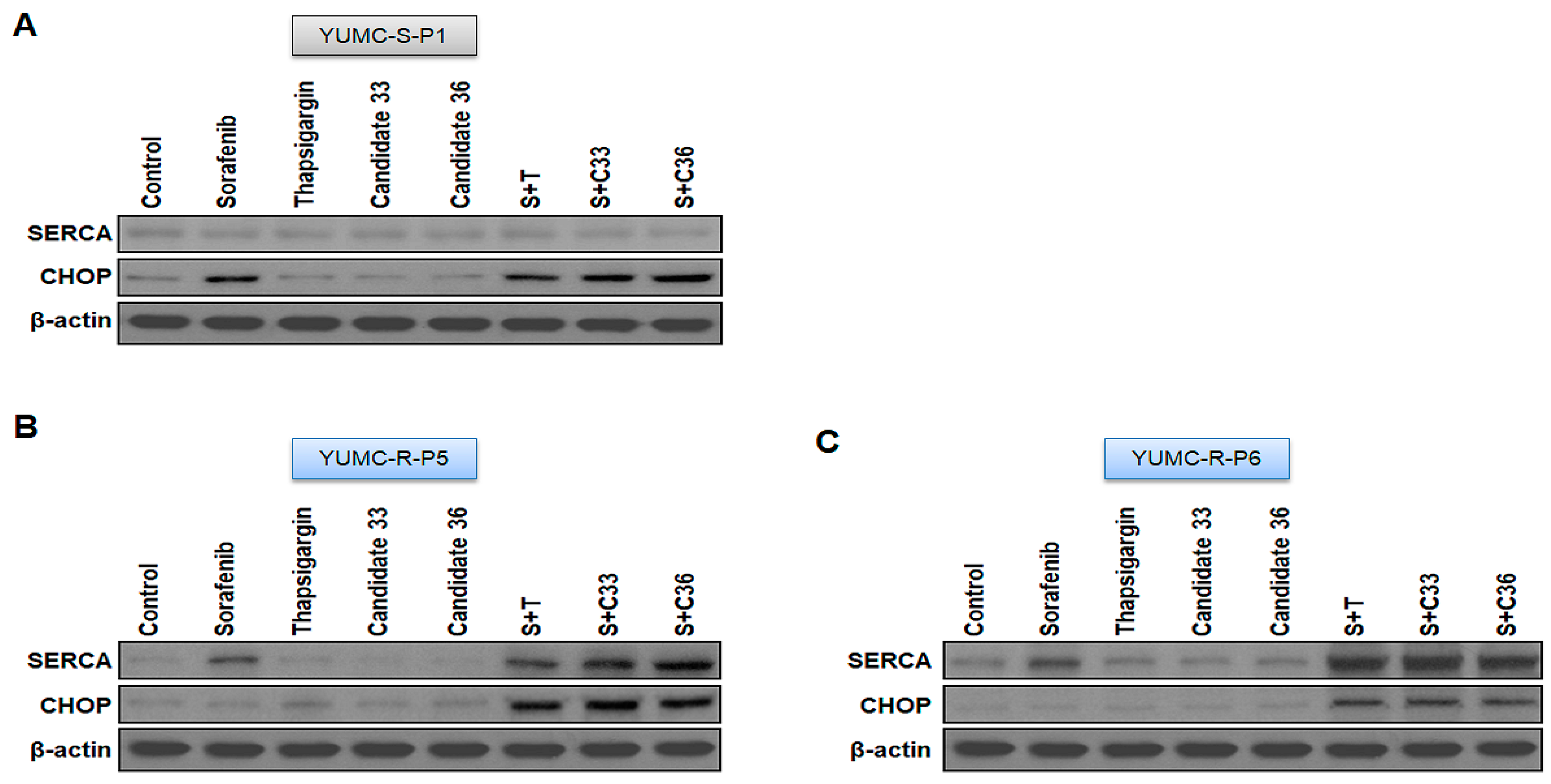
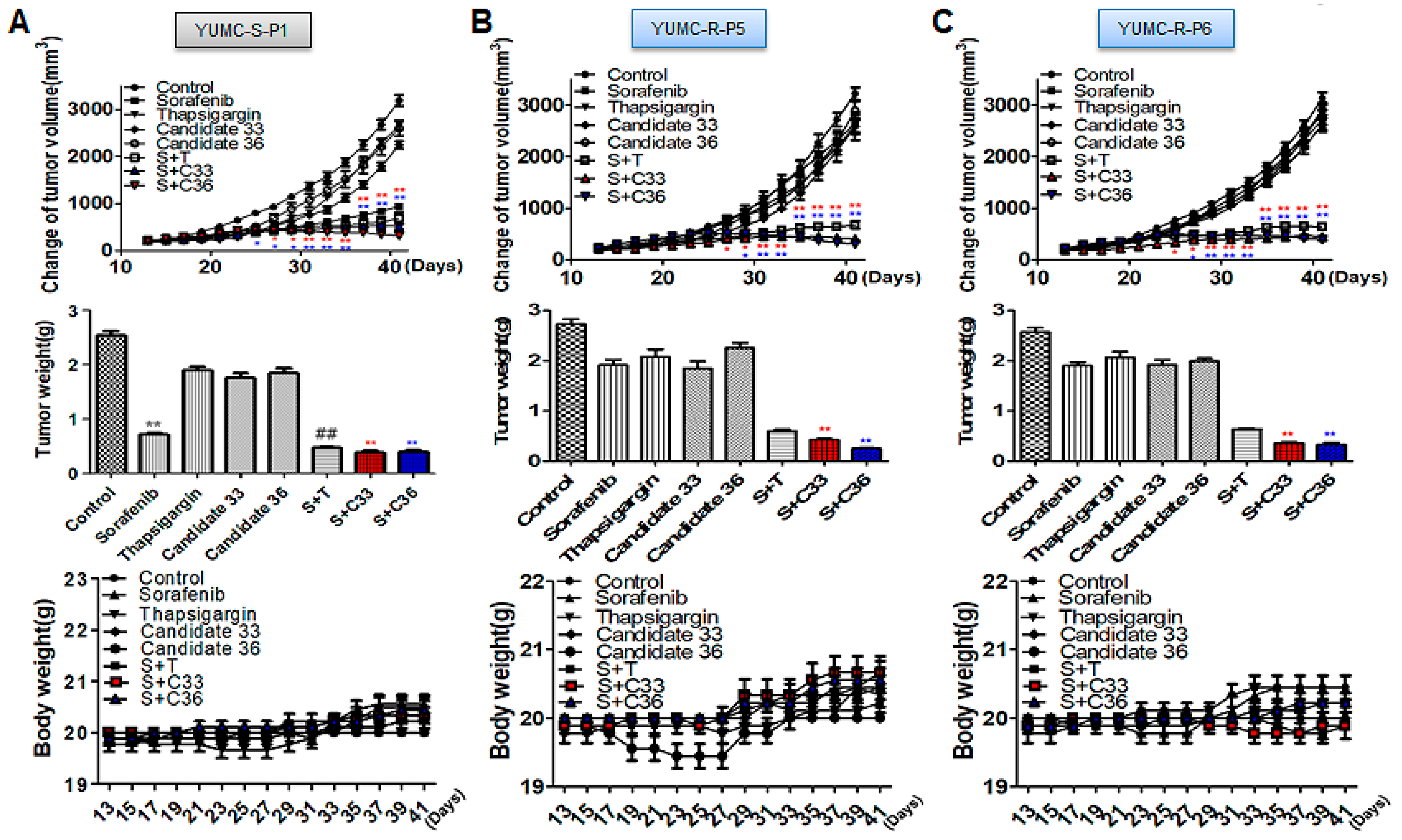
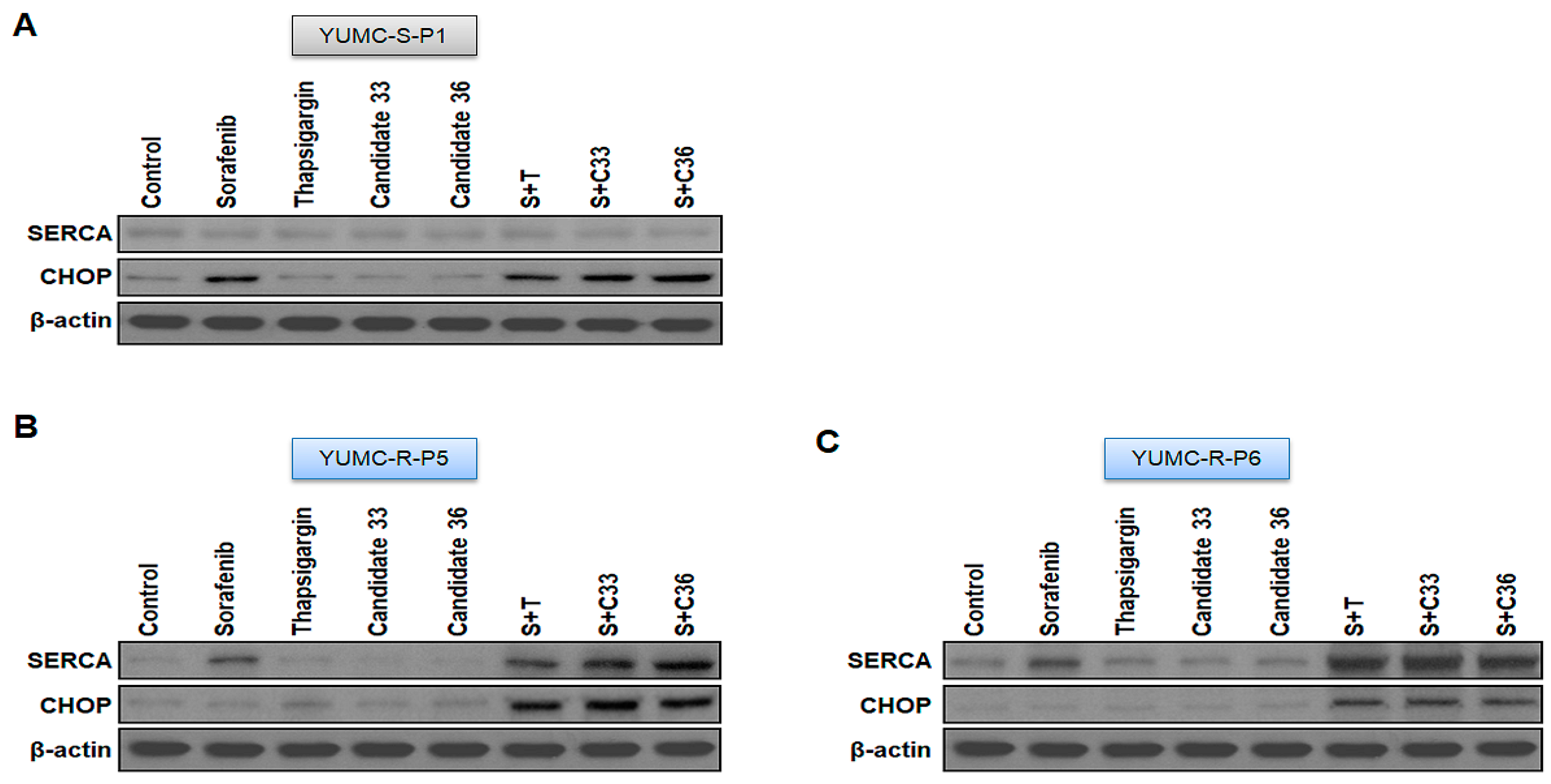
2.5. New Clinical Approach for Targeted Therapy through Novel Candidates 33 and 36 in a Patient-Derived Sorafenib-Resistant PTC Cell Mouse Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Study Design and Ethical Considerations
4.2. Patients
4.2.1. Patient 1
4.2.2. Patient 2 and 3
4.3. Patient Tissue Specimens
4.4. Primary Culture and Cancer Cell Isolation
4.5. mRNA-Seq Data
4.6. Statistical Analysis of Gene Expression Level
4.7. Hierarchical Clustering
4.8. Cell Viability Assay
4.9. Immunoblot Analysis
4.10. Used Information of Calcium Channel, NCX, PMCA and SERCA Inhibitors
4.11. Human PTC Cell Xenograft
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, Q.T.; Lee, E.J.; Huang, M.G.; Park, Y.I.; Khullar, A.; Plodkowski, R.A. Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits 2015, 8, 30–40. [Google Scholar] [PubMed]
- Fahiminiya, S.; de Kock, L.; Foulkes, W.D. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 2306–2307. [Google Scholar] [PubMed]
- Owonikoko, T.K.; Chowdry, R.P.; Chen, Z.; Kim, S.; Saba, N.F.; Shin, D.M.; Khuri, F.R. Clinical efficacy of targeted biologic agents as second-line therapy of advanced thyroid cancer. Oncologist 2013, 18, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Lim, J.H.; Kim, S.Y.; Kim, S.M.; Park, K.C. Discovery of pharmaceutical composition for prevention and treatment in patient-derived metastatic medullary thyroid carcinoma model. Biomedicines 2022, 10, 1901. [Google Scholar] [CrossRef]
- Raue, F.; Frank-Raue, K. Thyroid cancer: Risk-stratified management and individualized therapy. Clin. Cancer Res. 2016, 22, 5012–5021. [Google Scholar] [CrossRef]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly differentiated carcinoma of the thyroid gland: Current status and future prospects. Thyroid 2019, 29, 311–321. [Google Scholar] [CrossRef]
- Alwelaie, Y.; Howaidi, A.; Tashkandi, M.; Almotairi, A.; Saied, H.; Muzzaffar, M.; Alghamdi, D. Revisiting the cytomorphological features of poorly differentiated thyroid carcinoma: A comparative analysis with indeterminate thyroid fine-needle aspiration samples. J. Am. Soc. Cytopathol. 2023, 12, 331–340. [Google Scholar] [CrossRef]
- Kurczyk, A.; Gawin, M.; Chekan, M.; Wilk, A.; Lakomiec, K.; Mrukwa, G.; Fratczak, K.; Polanska, J.; Fujarewicz, K.; Pietrowska, M.; et al. Classification of thyroid tumors based on mass spectrometry imaging of tissue microarrays; a single-pixel approach. Int. J. Mol. Sci. 2020, 21, 6289. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, Z.; Liu, T.; Tang, M.; Mi, L.; Zhu, J.; Wu, W.; Wei, T. Targeted therapy and drug resistance in thyroid cancer. Eur. J. Med. Chem. 2022, 238, 114500. [Google Scholar] [CrossRef]
- Stassi, G.; Todaro, M.; Zerilli, M.; Ricci-Vitiani, L.; Di Liberto, D.; Patti, M.; Florena, A.; Di Gaudio, F.; Di Gesu, G.; De Maria, R. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res. 2003, 63, 6784–6790. [Google Scholar]
- Kim, B.H.; Kim, I.J.; Lee, B.J.; Lee, J.C.; Kim, I.S.; Kim, S.J.; Kim, W.J.; Jeon, Y.K.; Kim, S.S.; Kim, Y.K. Detection of plasma braf(v600e) mutation is associated with lung metastasis in papillary thyroid carcinomas. Yonsei Med. J. 2015, 56, 634–640. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of parp inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Yu, M.; Yin, D.; Sun, D.; Zhu, Y.; Bu, Y.; Sang, M. Impact of runx2 on drug-resistant human pancreatic cancer cells with p53 mutations. BMC Cancer 2018, 18, 309. [Google Scholar] [CrossRef] [PubMed]
- He, Y. Systematic response of staurosporine scaffold-based inhibitors to drug-resistant cancer kinase mutations. Arch. Pharm. 2020, 353, e1900320. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (dcc-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant kit and pdgfra variants. Cancer Cell 2019, 35, 738–751 e739. [Google Scholar] [CrossRef]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updat. 2020, 49, 100671. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. Egfr-tkis resistance via egfr-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef]
- Baloch, Z.W.; Asa, S.L.; Barletta, J.A.; Ghossein, R.A.; Juhlin, C.C.; Jung, C.K.; LiVolsi, V.A.; Papotti, M.G.; Sobrinho-Simoes, M.; Tallini, G.; et al. Overview of the 2022 who classification of thyroid neoplasms. Endocr. Pathol. 2022, 33, 27–63. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Lin, Y.; Liang, J. Radioactive iodine-refractory differentiated thyroid cancer and redifferentiation therapy. Endocrinol. Metab. 2019, 34, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with braf(v600e)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Miftari, R.; Topciu, V.; Nura, A.; Haxhibeqiri, V. Management of the patient with aggressive and resistant papillary thyroid carcinoma. Med. Arch. 2016, 70, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. The serca pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. Serca control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Park, K.C.; Kim, J.M.; Kim, S.Y.; Kim, S.M.; Lim, J.H.; Kim, M.K.; Fang, S.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Pmca inhibition reverses drug resistance in clinically refractory cancer patient-derived models. BMC Med. 2023, 21, 38. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, K.; Lim, J.H.; Yun, H.J.; Kim, S.Y.; Choi, K.H.; Kim, C.W.; Lee, J.H.; Weicker, R.; Pan, C.H.; et al. Potential therapeutic agents against paclitaxel-and sorafenib-resistant papillary thyroid carcinoma. Int. J. Mol. Sci. 2022, 23, 10378. [Google Scholar] [CrossRef]
- Lim, J.H.; Choi, K.H.; Kim, S.Y.; Park, C.S.; Kim, S.M.; Park, K.C. Patient-derived, drug-resistant colon cancer cells evade chemotherapeutic drug effects via the induction of epithelial-mesenchymal transition-mediated angiogenesis. Int. J. Mol. Sci. 2020, 21, 7469. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Survival of cancer stem-like cells under metabolic stress via camk2alpha-mediated upregulation of sarco/endoplasmic reticulum calcium atpase expression. Clin. Cancer Res. 2018, 24, 1677–1690. [Google Scholar] [CrossRef]
- Pecina-Slaus, N. Wnt signal transduction pathway and apoptosis: A review. Cancer Cell Int. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Dev, A., Jr.; Vachher, M.; Prasad, C.P. Beta-catenin inhibitors in cancer therapeutics: Intricacies and way forward. Bioengineered 2023, 14, 2251696. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, A.A.; Benaim, G. [Ca2+ and sphingolipids as modulators for apoptosis and cancer]. Investig. Clin. 2012, 53, 84–110. [Google Scholar] [PubMed]
- Seo, J.A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ atpase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Lim, J.H.; Park, K.; Choi, K.H.; Kim, C.W.; Lee, J.H.; Weicker, R.; Pan, C.H.; Kim, S.M.; Park, K.C. Drug discovery using evolutionary similarities in chemical binding to inhibit patient-derived hepatocellular carcinoma. Int. J. Mol. Sci. 2022, 23, 7971. [Google Scholar] [CrossRef]
- Bergdorf, K.; Ferguson, D.C.; Mehrad, M.; Ely, K.; Stricker, T.; Weiss, V.L. Papillary thyroid carcinoma behavior: Clues in the tumor microenvironment. Endocr. Relat. Cancer 2019, 26, 601–614. [Google Scholar] [CrossRef]
- Lewinski, A.; Adamczewski, Z. Papillary thyroid carcinoma: A cancer with an extremely diverse genetic background and prognosis. Pol. Arch. Intern. Med. 2017, 127, 388–389. [Google Scholar] [CrossRef]
- Colombo, C.; Minna, E.; Gargiuli, C.; Muzza, M.; Dugo, M.; De Cecco, L.; Pogliaghi, G.; Tosi, D.; Bulfamante, G.; Greco, A.; et al. The molecular and gene/mirna expression profiles of radioiodine resistant papillary thyroid cancer. J. Exp. Clin. Cancer Res. 2020, 39, 245. [Google Scholar] [CrossRef]
- Zelinskaya, A. Immunocytochemical characteristics of thyrocytes in radioiodine refractory metastases of papillary thyroid cancer. Exp. Oncol. 2019, 41, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, R.; Adamo, L.; Iannolo, G.; Vicari, L.; Giuffrida, D.; Eramo, A.; Gulisano, M.; Memeo, L.; Conticello, C. Resistance of papillary thyroid cancer stem cells to chemotherapy. Oncol. Lett. 2016, 12, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef]
- Davies, L.; Welch, H.G. Increasing incidence of thyroid cancer in the united states, 1973–2002. JAMA 2006, 295, 2164–2167. [Google Scholar] [CrossRef]
- De Groot, J.W.; Links, T.P.; Plukker, J.T.; Lips, C.J.; Hofstra, R.M. Ret as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr. Rev. 2006, 27, 535–560. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Marlow, L.A.; Copland, J.A. Anaplastic thyroid cancer: Molecular pathogenesis and emerging therapies. Endocr. Relat. Cancer 2009, 16, 17–44. [Google Scholar] [CrossRef]
- Ivan, M.; Bond, J.A.; Prat, M.; Comoglio, P.M.; Wynford-Thomas, D. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene 1997, 14, 2417–2423. [Google Scholar] [CrossRef]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and neoadjuvant breast cancer treatments: A systematic review of their effects on mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Sturm, N.; Guthle, M.; Huttner, F.J.; Perkhofer, L. Pancreatic cancer: Current multimodality treatment options and the future impact of molecular biological profiling. Visc. Med. 2022, 38, 20–29. [Google Scholar] [CrossRef]
- Heinemann, V.; Stintzing, S. [Neoadjuvant and adjuvant therapy of resectable colon cancer—Current standards and developments]. Dtsch. Med. Wochenschr. 2021, 146, 1457–1467. [Google Scholar] [PubMed]
- Friedlaender, A.; Naidoo, J.; Banna, G.L.; Metro, G.; Forde, P.; Addeo, A. Role and impact of immune checkpoint inhibitors in neoadjuvant treatment for nsclc. Cancer Treat. Rev. 2022, 104, 102350. [Google Scholar] [CrossRef] [PubMed]
- Akateh, C.; Black, S.M.; Conteh, L.; Miller, E.D.; Noonan, A.; Elliott, E.; Pawlik, T.M.; Tsung, A.; Cloyd, J.M. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3704–3721. [Google Scholar] [CrossRef]
- Foerster, F.; Galle, P.R. The current landscape of clinical trials for systemic treatment of hcc. Cancers 2021, 13, 1962. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Khalil, L.; Millett, R.; Kaseb, A. Neoadjuvant and adjuvant treatment approaches for hepatocellular carcinoma: Future outlook. Chin. Clin. Oncol. 2021, 10, 7. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Xavier, C.P.; Pesic, M.; Vasconcelos, M.H. Understanding cancer drug resistance by developing and studying resistant cell line models. Curr. Cancer Drug Targets 2016, 16, 226–237. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (emt) to overcome drug resistance in cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Alasiri, G.; Jiramongkol, Y.; Zona, S.; Fan, L.Y.; Mahmud, Z.; Gong, G.; Lee, H.J.; Lam, E.W. Regulation of perk expression by foxo3: A vulnerability of drug-resistant cancer cells. Oncogene 2019, 38, 6382–6398. [Google Scholar] [CrossRef]
- Fernandes, C.; Prabhu, P.; Juvale, K.; Suares, D.; Yc, M. Cancer cell fusion: A potential target to tackle drug-resistant and metastatic cancer cells. Drug Discov. Today 2019, 24, 1836–1844. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.R.; Etherington, M.D.; Salmon, G.P.; McNally, S. The influence of nicardipine on platelet tests in patients with claudication. Thromb. Res. 1990, 58, 75–80. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. Mcur1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ros/nrf2/notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef]
- Sun, J.; Ailiman, M. Regulation of calcium pump through notch/jagged/hes signaling pathway in canine model of chronic atrial fibrillation. Int. J. Clin. Exp. Pathol. 2019, 12, 4034–4040. [Google Scholar]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic notch signaling with serca inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef]
- Marchesini, M.; Gherli, A.; Montanaro, A.; Patrizi, L.; Sorrentino, C.; Pagliaro, L.; Rompietti, C.; Kitara, S.; Heit, S.; Olesen, C.E.; et al. Blockade of oncogenic notch1 with the serca inhibitor cad204520 in t cell acute lymphoblastic leukemia. Cell Chem. Biol. 2020, 27, 678–697.e13. [Google Scholar] [CrossRef]
- Chang, H.S.; Kim, Y.; Lee, S.Y.; Yun, H.J.; Chang, H.J.; Park, K.C. Anti-cancer serca inhibitors targeting sorafenib-resistant human papillary thyroid carcinoma. Int. J. Mol. Sci. 2023, 24, 7069. [Google Scholar] [CrossRef]
- Yun, H.J.; Kim, M.; Kim, S.Y.; Fang, S.; Kim, Y.; Chang, H.S.; Chang, H.J.; Park, K.C. Effects of anti-cancer drug sensitivity-related genetic differences on therapeutic approaches in refractory papillary thyroid cancer. Int. J. Mol. Sci. 2022, 23, 699. [Google Scholar] [CrossRef]
- Kim, N.; Kim, K.H.; Lim, W.J.; Kim, J.; Kim, S.A.; Yoo, H.J. Whole exome sequencing identifies novel de novo variants interacting with six gene networks in autism spectrum disorder. Genes 2020, 12, 1. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| YUMC-S-P1 | YUMC-R-P5 | YUMC-R-P6 | |
|---|---|---|---|
| Age at Diagnosis | 53 | 52 | 57 |
| Gender | Male | Female | Male |
| Primary Disease Site | Thyroid | Thyroid | Thyroid |
| Stage | T4aN1bM0 | T4aN1bM1 | T3N1bM1 |
| Primary Pathology | Papillary thyroid cancer | Papillary thyroid cancer (Recurrence and metastasis after sorafenib treatment) | Papillary thyroid cancer (Recurrence and metastasis after sorafenib treatment) |
| Classification of specimen used for culture | Fresh tumor | Fresh tumor | Fresh tumor |
| Obtained from | Severance Hospital, Seoul, Republic of Korea | Severance Hospital, Seoul, Republic of Korea | Severance Hospital, Seoul, Republic of Korea |
| Cell Line | Histopathology | Animal | Cell Proliferation IC50 (μM) | |||
|---|---|---|---|---|---|---|
| Sorafenib | S + T | S + C33 | S + C36 | |||
| YUMC-S-P1 | Thyroid, Papillary | Human | 22 (±0.2) | 22 (±0.3) | 22 (±0.1) | 22 (±0.1) |
| YUMC-R-P5 | Thyroid, Papillary | Human | – | 22 (±0.1) | 23 (±0.1) | 23 (±0.3) |
| YUMC-R-P6 | Thyroid, Papillary | Human | – | 22 (±0.3) | 23 (±0.2) | 23 (±0.1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Yun, H.J.; Choi, K.H.; Kim, C.W.; Lee, J.H.; Weicker, R.; Kim, S.-M.; Park, K.C. Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 16413. https://doi.org/10.3390/ijms242216413
Kim Y, Yun HJ, Choi KH, Kim CW, Lee JH, Weicker R, Kim S-M, Park KC. Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer. International Journal of Molecular Sciences. 2023; 24(22):16413. https://doi.org/10.3390/ijms242216413
Chicago/Turabian StyleKim, Yuna, Hyeok Jun Yun, Kyung Hwa Choi, Chan Wung Kim, Jae Ha Lee, Raymond Weicker, Seok-Mo Kim, and Ki Cheong Park. 2023. "Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer" International Journal of Molecular Sciences 24, no. 22: 16413. https://doi.org/10.3390/ijms242216413
APA StyleKim, Y., Yun, H. J., Choi, K. H., Kim, C. W., Lee, J. H., Weicker, R., Kim, S.-M., & Park, K. C. (2023). Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer. International Journal of Molecular Sciences, 24(22), 16413. https://doi.org/10.3390/ijms242216413