

PHB2 Alleviates Neurotoxicity of Prion Peptide PrP106–126 via PINK1/Parkin-Dependent Mitophagy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
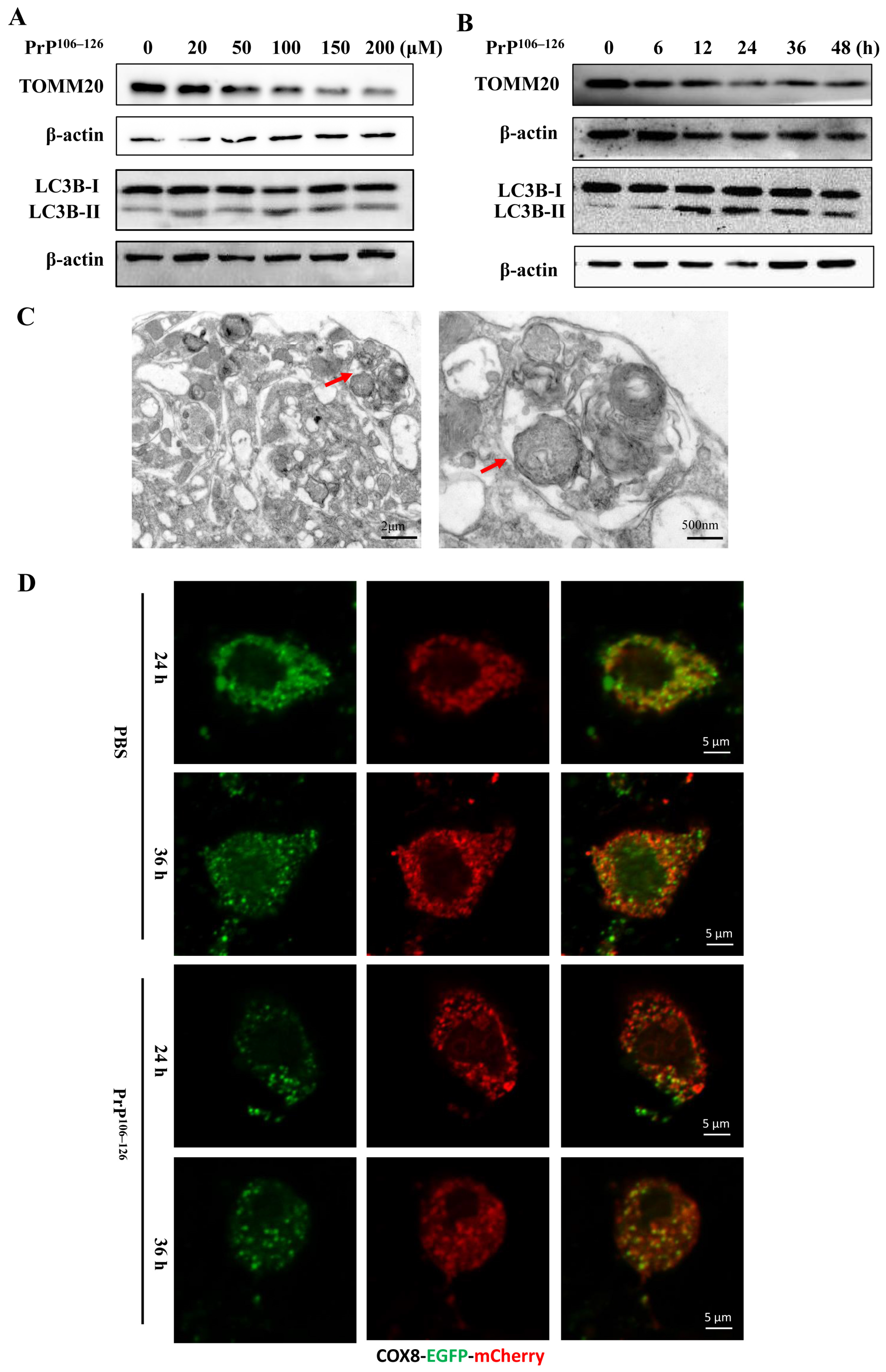
2.1. PrP106–126 Caused Morphological Abnormality of Mitochondria in the Primary Neurons
2.2. PrP106–126 Induced Mitophagy in Primary Neurons
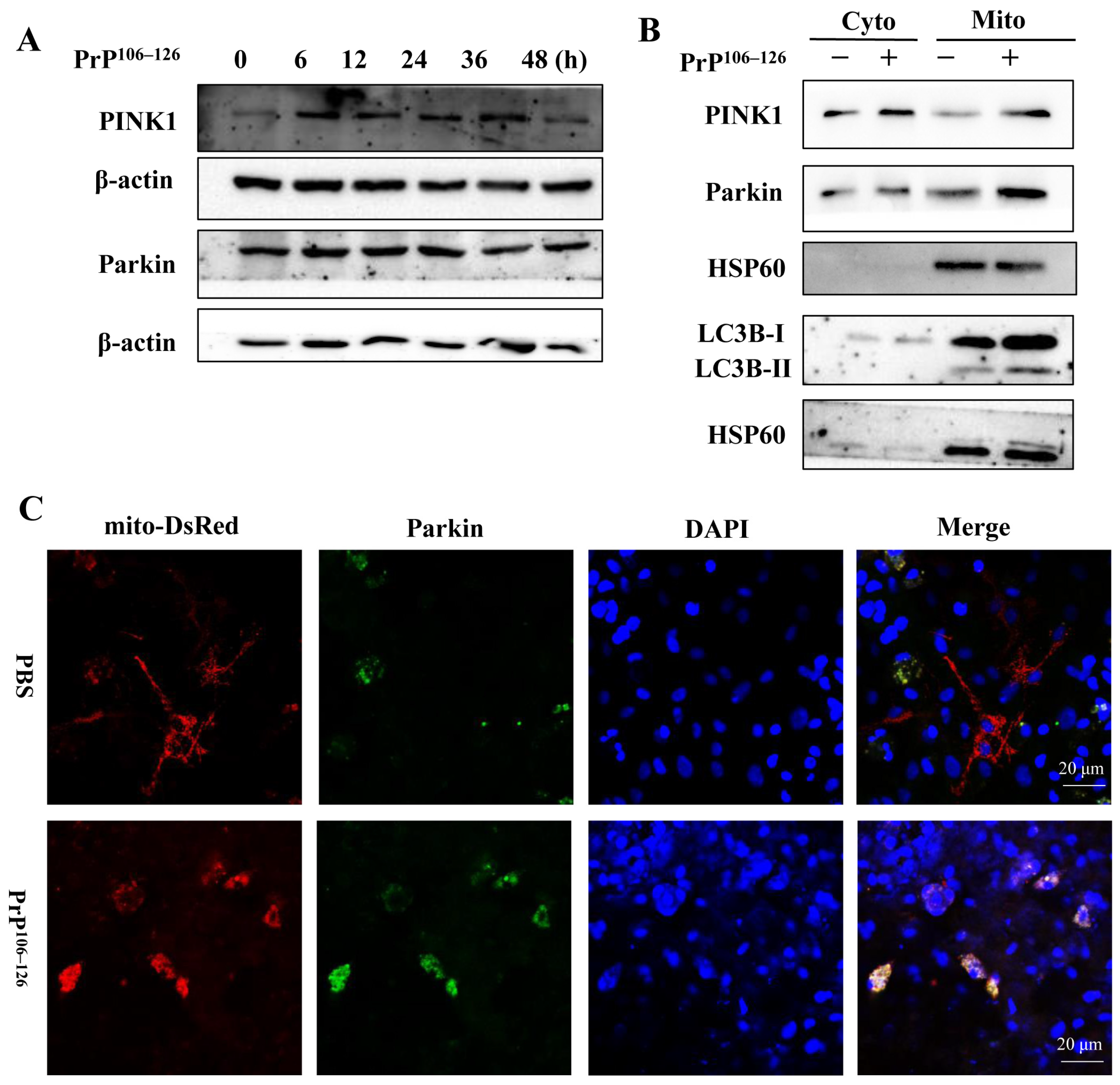
2.3. PrP106–126 Triggers PINK1/Parkin-Dependent Mitophagy
2.4. PHB2 Involves in PrP106–126-Induced Mitophagy in Primary Neurons
2.5. PHB2 Is Required for PINK1/Parkin-Dependent Mitophagy in PrP106–126-Treated Neurons
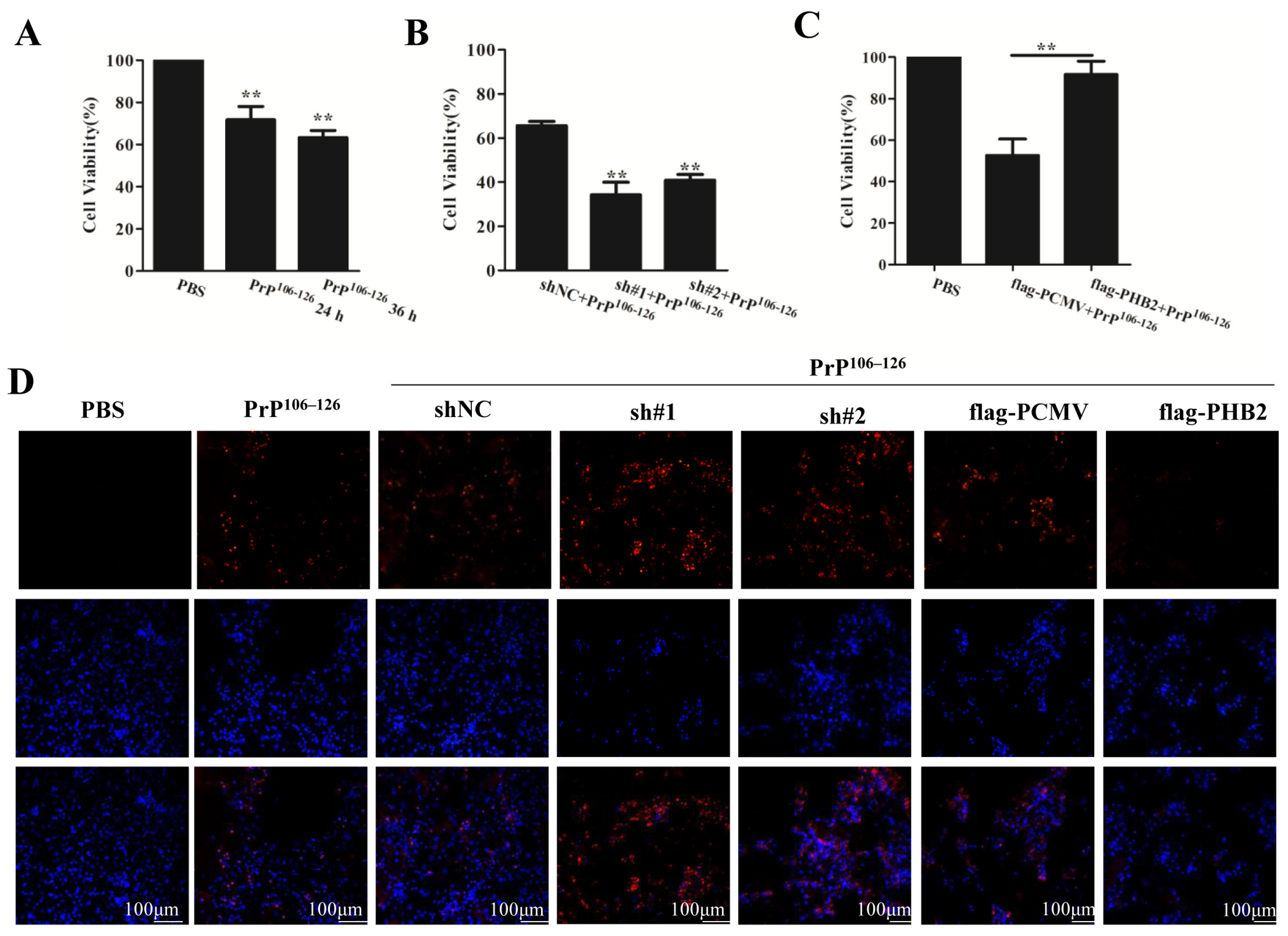
2.6. PHB2 Modulates PrP106–126-Induced Neuronal Death
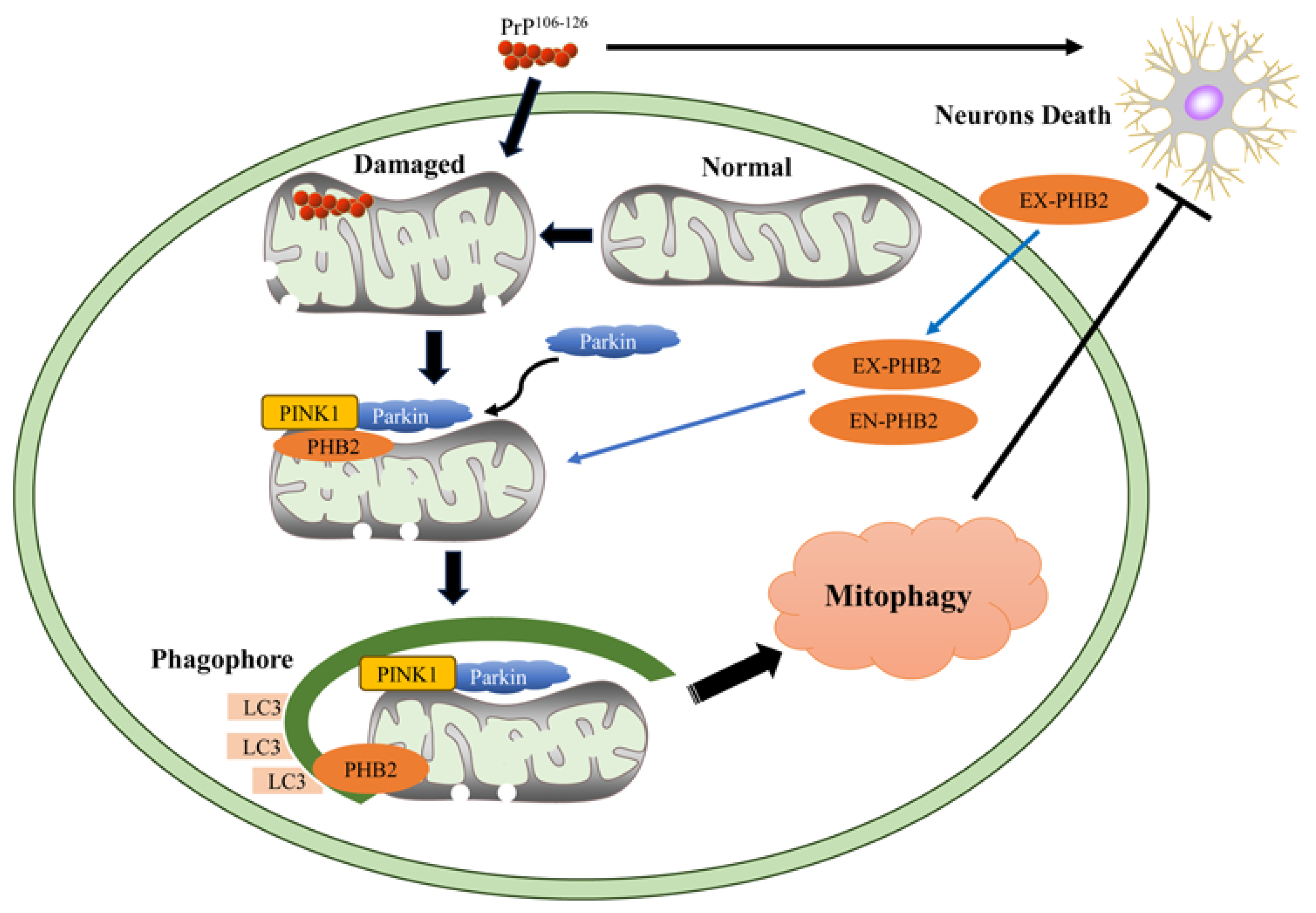
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Prion Protein Peptide and Peptide Treatment
4.4. Mitochondrial Isolation
4.5. Immunofluorescence Microscopy
4.6. Plasmids and Transfection
4.7. Western Blotting Analysis
4.8. Transmission Electron Microscopy (TEM)
4.9. Cell Viability Assay
4.10. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviations | Full Name |
| PrP106–126 | PrP fragment 106–126 |
| CNS | Central Nervous System |
| PINK1 | PTEN-induced kinase 1 |
| PHB2 | Prohibitin 2 |
| TOMM20 | Translocase of the Outer Mitochondrial Membrane 20 |
| LC3B | Light Chain 3B |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| PBS | Phosphate Buffer Saline |
| FBS | Fetal Bovine Serum |
| HBSS | Hank’s Balanced Salt Solution |
| SDS-PAGE | Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis |
| DAPI | 4,6-diamidina-2-phenylin |
| shRNA | Short hairpin RNA |
| TEM | Transmission Electron Microscopy |
| CCK | Cell Counting Kit |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| PI | Propidium Iodide |
| ANOVA | Analysis of Variance |
| SD | Standard Deviation |
| SPSS | Statistical Product and Service Solutions |
References
- Bradford, B.M.; Mabbott, N.A. Prion disease and the innate immune system. Viruses 2012, 4, 3389–3419. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, N.S.; Sakasegawa, Y.; Kaneko, K. Therapeutic Approaches in Prion Disease. J. Health Sci. 2003, 49, 267–272. [Google Scholar] [CrossRef][Green Version]
- Fang, C.; Imberdis, T.; Garza, M.C.; Wille, H.; Harris, D.A. A Neuronal Culture System to Detect Prion Synaptotoxicity. PLoS Pathog. 2016, 12, e1005623. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Nobel Lecture: Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S. Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Klemm, H.M.; Welton, J.M.; Masters, C.L.; Klug, G.M.; Boyd, A.; Hill, A.F.; Collins, S.J.; Lawson, V.A. The prion protein preference of sporadic Creutzfeldt-Jakob disease subtypes. J. Biol. Chem. 2012, 287, 36465–36472. [Google Scholar] [CrossRef]
- Collins, C.L. Transmissible spongiform encephalopathies. Lancet 2004, 363, 51–61. [Google Scholar] [CrossRef]
- Li, C.; Wang, D.; Wu, W.; Yang, W.; Shah SZ, A.; Zhao, Y.; Duan, Y.; Wang, L.; Zhou, X.; Zhao, D.; et al. DLP1-dependent mitochondrial fragmentation and redistribution mediate prion-associated mitochondrial dysfunction and neuronal death. Aging Cell 2018, 17, e12693. [Google Scholar] [CrossRef]
- Forloni, G.; Chiesa, R.; Bugiani, O.; Salmona, M.; Tagliavini, F. Review: PrP 106-126—25 years after. Neuropathol. Appl. Neurobiol. 2019, 45, 430–440. [Google Scholar] [CrossRef]
- Jeong, J.K.; Lee, J.H.; Moon, J.H.; Lee, Y.J.; Park, S.Y. Melatonin-mediated β-catenin activation protects neuron cells against prion protein-induced neurotoxicity. J. Pineal Res. 2014, 57, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Finkel, E. The mitochondrion: Is it central to apoptosis? Science 2001, 292, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.P.; Xiao, K.; Wu, Y.Z.; Chen, D.D.; Yang, X.H.; Shi, Q.; Dong, X.P. Enhanced Mitophagy Activity in Prion-Infected Cultured Cells and Prion-Infected Experimental Mice via a Pink1/Parkin-Dependent Mitophagy Pathway. ACS Chem. Neurosci. 2020, 11, 814–829. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Choi, Y.G.; Shin, H.Y.; Oh, J.M.; Park, J.H.; Kim, J.T.; Carp, R.I.; Choi, E.K.; Kim, Y.S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014, 448, 157–162. [Google Scholar] [CrossRef]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, Y.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 2012, 1833, 205–212. [Google Scholar] [CrossRef]
- Dieter, A.K.; Asa, B.G. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar]
- Zhu, T.; Chen, J.L.; Wang, Q.; Shao, W.; Qi, B. Modulation of Mitochondrial Dynamics in Neurodegenerative Diseases: An Insight Into Prion Diseases. Front. Aging Neurosci. 2018, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J.; Green, D.R. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; Lavoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhou, Y.; Lu, Y.; Zhou, K.; Cai, W. PHB2 interacts with LC3 and SQSTM1 is required for bile acids-induced mitophagy in cholestatic liver. Cell Death Dis. 2018, 9, 160. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Fiskum, G. Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 2004, 90, 1281–1289. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641. [Google Scholar] [CrossRef] [PubMed]
- Aiken, J.M.; Williamson, J.L.; Marsh, R.F. Evidence of mitochondrial involvement in scrapie infection. J. Virol. 1989, 63, 1686–1694. [Google Scholar] [CrossRef]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Désaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2019, 16, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hou, H.; Ma, G.; Ma, Q.; Li, N.; Zhang, L.; Dong, C.; Cao, M.; Tam, K.Y.; Ying, Z.; et al. The interaction between E3 ubiquitin ligase Parkin and mitophagy receptor PHB2 links inner mitochondrial membrane ubiquitination to efficient mitophagy. J. Biol. Chem. 2022, 298, 102704. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Kretzschmar, H.A. Prion-induced neuronal damage—The mechanisms of neuronal destruction in the subacute spongiform encephalopathies. Curr. Top. Microbiol. Immunol. 2001, 253, 203–217. [Google Scholar]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Arch. Eur. J. Physiol. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Ju, W.K.; Choi, E.K.; Kim, J.; Lea, H.Z.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. 1998, 96, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Binyamin, O.; Frid, K.; Saada, A.; Gabizon, R. Mitochondrial dysfunction in preclinical genetic prion disease: A target for preventive treatment? Neurobiol. Dis. 2018, 124, 57–66. [Google Scholar] [CrossRef]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial Alterations near Amyloid Plaques in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef]
- Zhang, L.; Dai, L.; Li, D. Mitophagy in neurological disorders. J. Neuroinflamm. 2021, 18, 297. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef]
- Valentina, S.; Lorenzo, G.; José, M.B.-S.P.; Valentina, I.; Chiara, M.M.; Guido, K. Organelle-Specific Initiation of Autophagy. Mol. Cell 2015, 59, 522–539. [Google Scholar]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.X.; Correia, S.C.; Carvalho, C.; Cardoso, S.; Santos, M.S.; Moreira, P.I. Mitophagy in Neurodegeneration: An Opportunity for Therapy? Curr. Drug Targets 2011, 12, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Chen, Y.; Xu, X.; Wu, R.; He, F.; Zhao, Q.; Sun, Q.; Yi, C.; Wu, J.; Najafov, A.; et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat. Commun. 2020, 11, 5731. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; Mcbride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2014, 6, 871–878. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Springer, W.; Kahle, P.J. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011, 7, 266–278. [Google Scholar] [CrossRef]
- Wang, L.; Tang, X.Q.; Shi, Y.; Li, H.M.; Meng, Z.Y.; Chen, H.; Li, X.H.; Chen, Y.C.; Liu, H.; Hong, Y.; et al. Tetrahydroberberrubine retards heart aging in mice by promoting PHB2-mediated mitophagy. Acta Pharmacol. Sin. 2023, 44, 332–344. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Prohibitins. Curr. Biol. 2017, 27, R629–R631. [Google Scholar] [CrossRef]
- Nijtmans, L.G.J.; Artal, S.M.; Grivell, L.A.; Coates, P.J. The mitochondrial PHB complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell. Mol. Life Sci. CMLS 2002, 59, 143–155. [Google Scholar] [CrossRef]
- He, L.; Zhou, H.; Liu, H.; Qu, H. Prohibitin 2/PHB2 in Parkin-mediated mitophagy: A potential therapeutic target for mitochondrial diseases. Acta Biochim. Biophys. Sin. 2017, 49, 750–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yin, C.; Liu, X.; Bai, X.; Wang, L.; Xu, H.; Ju, J.; Zhang, L. Prohibitin 2/PHB2 in Parkin-Mediated Mitophagy: A Potential Therapeutic Target for Non-Small Cell Lung Carcinoma. Med. Sci. Monit. 2020, 26, e923227. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, M.; Zhang, X.; Li, Z.; Yang, D.; Zhao, M.; Wang, D.; Sun, Z.; Ehsan, S.; Li, W.; et al. PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease. Cell Death Dis. 2022, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; Jin, W.; Shen, M.; Yin, S.; Lai, X.; Ma, H.; Jiang, M.; Sun, D.; Yan, J. Nonreceptor Tyrosine Kinase c-Abl-Mediated PHB2 Phosphorylation Aggravates Mitophagy Disorder in Parkinson’s Disease Model. Oxid. Med. Cell Longev. 2022, 2022, 9233749. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Zhang, Y.; Wu, J.; Shen, M.; Yin, S.; Yan, J. Rutin Attenuates Oxidative Stress Via PHB2-Mediated Mitophagy in MPP. Neurotox. Res. 2023, 41, 242–255. [Google Scholar] [CrossRef]
- Jiang, M.; Lai, X.; Zhang, Y.; Shen, M.; Ma, H.; Liu, A.; Wu, J.; Yan, J. Artemisinin Alleviates Cerebral Ischemia/Reperfusion-Induced Oxidative Damage via Regulating PHB2-Mediated Autophagy in the Human Neuroblastoma SH-SY5Y Cell Line. Oxid. Med. Cell Longev. 2022, 2022, 6568748. [Google Scholar] [CrossRef]
- Song, Z.; Shah, S.Z.A.; Yang, W.; Dong, H.; Yang, L.; Zhou, X.; Zhao, D. Downregulation of the Repressor Element 1-Silencing Transcription Factor (REST) Is Associated with Akt-mTOR and Wnt-beta-Catenin Signaling in Prion Diseases Models. Front. Mol. Neurosci. 2017, 10, 128. [Google Scholar] [CrossRef]
- Zhu, T.; Zhao, D.; Song, Z.; Yuan, Z.; Li, C.; Wang, Y.; Zhou, X.; Yin, X.; Hassan, M.F.; Yang, L. HDAC6 alleviates prion peptide-mediated neuronal death via modulating PI3K-Akt-mTOR pathway. Neurobiol. Aging 2016, 37, 91–102. [Google Scholar] [CrossRef]
- Song, Z.; Yang, L.; Wang, Y.; Zhu, T.; Zhou, X.; Yin, X.; Yao, H.; Zhao, D. Overexpression of BAT3 alleviates prion protein fragment PrP106–126-induced neuronal apoptosis. CNS Neurosci. Ther. 2014, 20, 737–747. [Google Scholar] [CrossRef]
- Yang, W.; Yang, L.F.; Song, Z.Q.; Shah, S.Z.A.; Cui, Y.Y.; Li, C.D.; Zhao, H.F.; Gao, H.L.; Zhou, X.M.; Zhao, D.M. PRAS40 alleviates neurotoxic prion peptide-induced apoptosis via mTOR-AKT signaling. CNS Neurosci. Ther. 2017, 23, 416–427. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Liu, K.; Xie, Q.; Xin, H.; Chen, W.; Lin, S.; Feng, D.; Zhu, T. PHB2 Alleviates Neurotoxicity of Prion Peptide PrP106–126 via PINK1/Parkin-Dependent Mitophagy. Int. J. Mol. Sci. 2023, 24, 15919. https://doi.org/10.3390/ijms242115919
Zheng X, Liu K, Xie Q, Xin H, Chen W, Lin S, Feng D, Zhu T. PHB2 Alleviates Neurotoxicity of Prion Peptide PrP106–126 via PINK1/Parkin-Dependent Mitophagy. International Journal of Molecular Sciences. 2023; 24(21):15919. https://doi.org/10.3390/ijms242115919
Chicago/Turabian StyleZheng, Xiaohui, Kun Liu, Qingqing Xie, Hangkuo Xin, Wei Chen, Shengyu Lin, Danqi Feng, and Ting Zhu. 2023. "PHB2 Alleviates Neurotoxicity of Prion Peptide PrP106–126 via PINK1/Parkin-Dependent Mitophagy" International Journal of Molecular Sciences 24, no. 21: 15919. https://doi.org/10.3390/ijms242115919
APA StyleZheng, X., Liu, K., Xie, Q., Xin, H., Chen, W., Lin, S., Feng, D., & Zhu, T. (2023). PHB2 Alleviates Neurotoxicity of Prion Peptide PrP106–126 via PINK1/Parkin-Dependent Mitophagy. International Journal of Molecular Sciences, 24(21), 15919. https://doi.org/10.3390/ijms242115919