
Cucurbitacin I Reverses Tumor-Associated Macrophage Polarization to Affect Cancer Cell Metastasis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
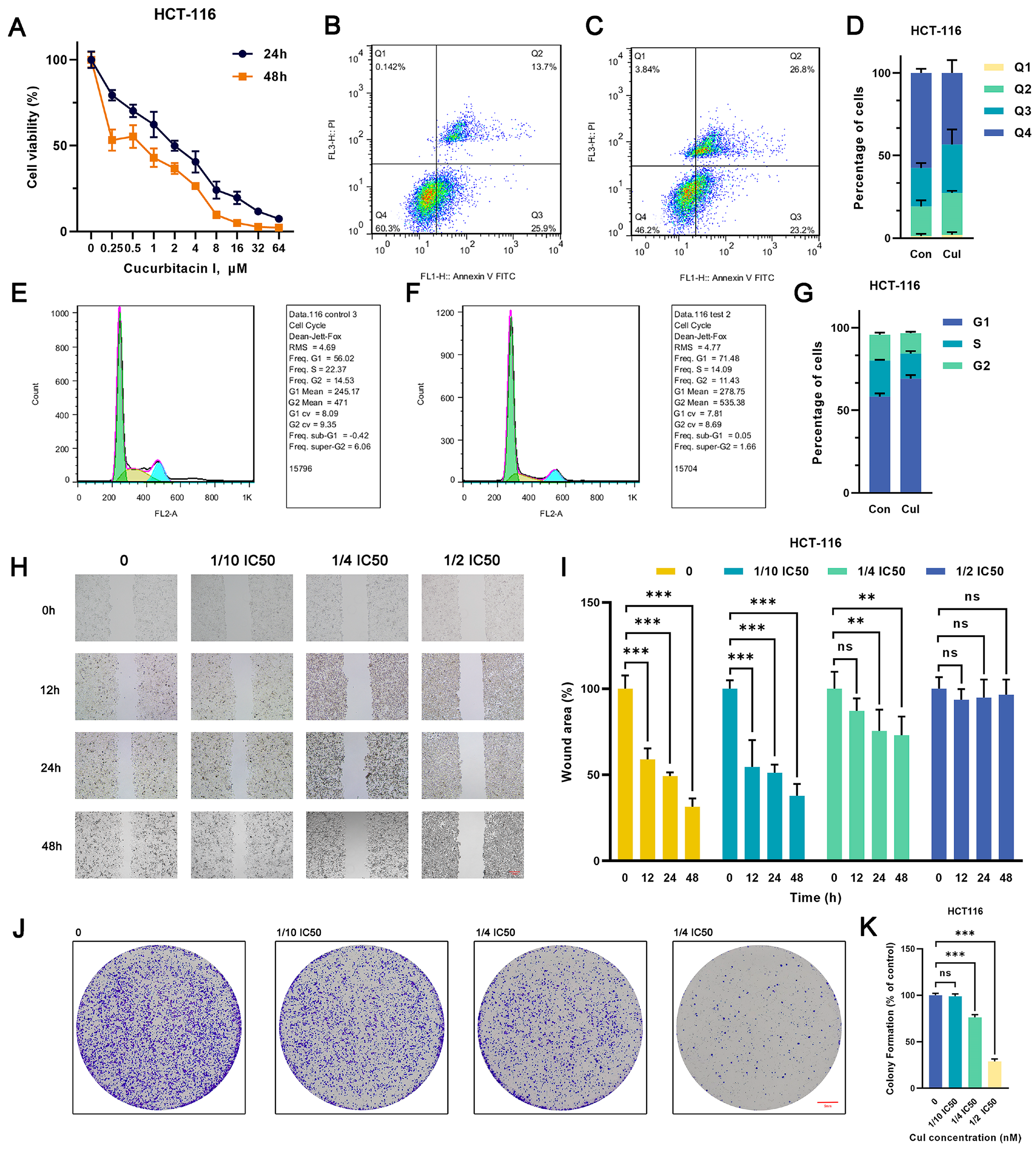
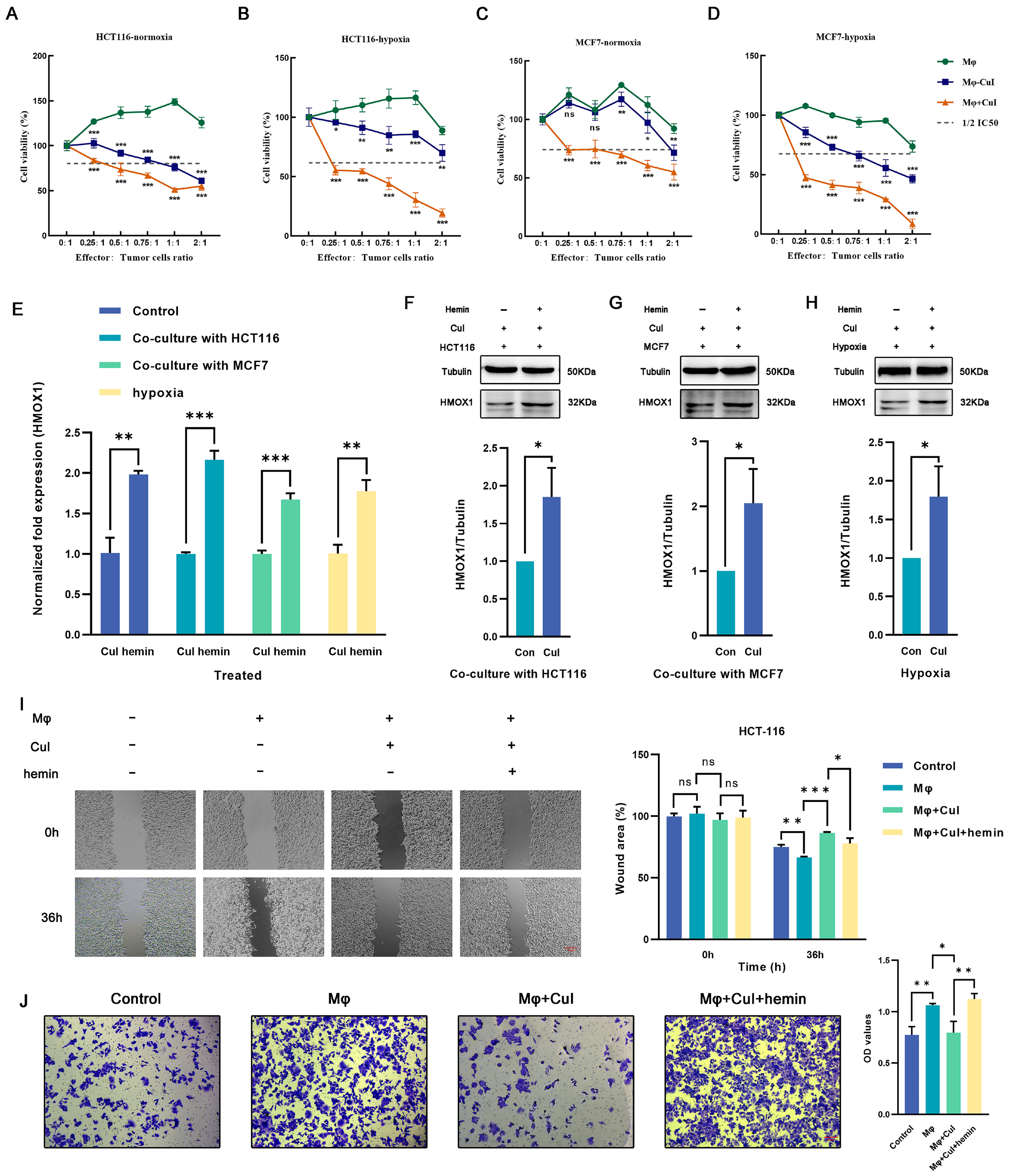
2.1. Cucurbitacin I Exerts Antitumorigenic Effects on Breast and Colorectal Cancer Cells
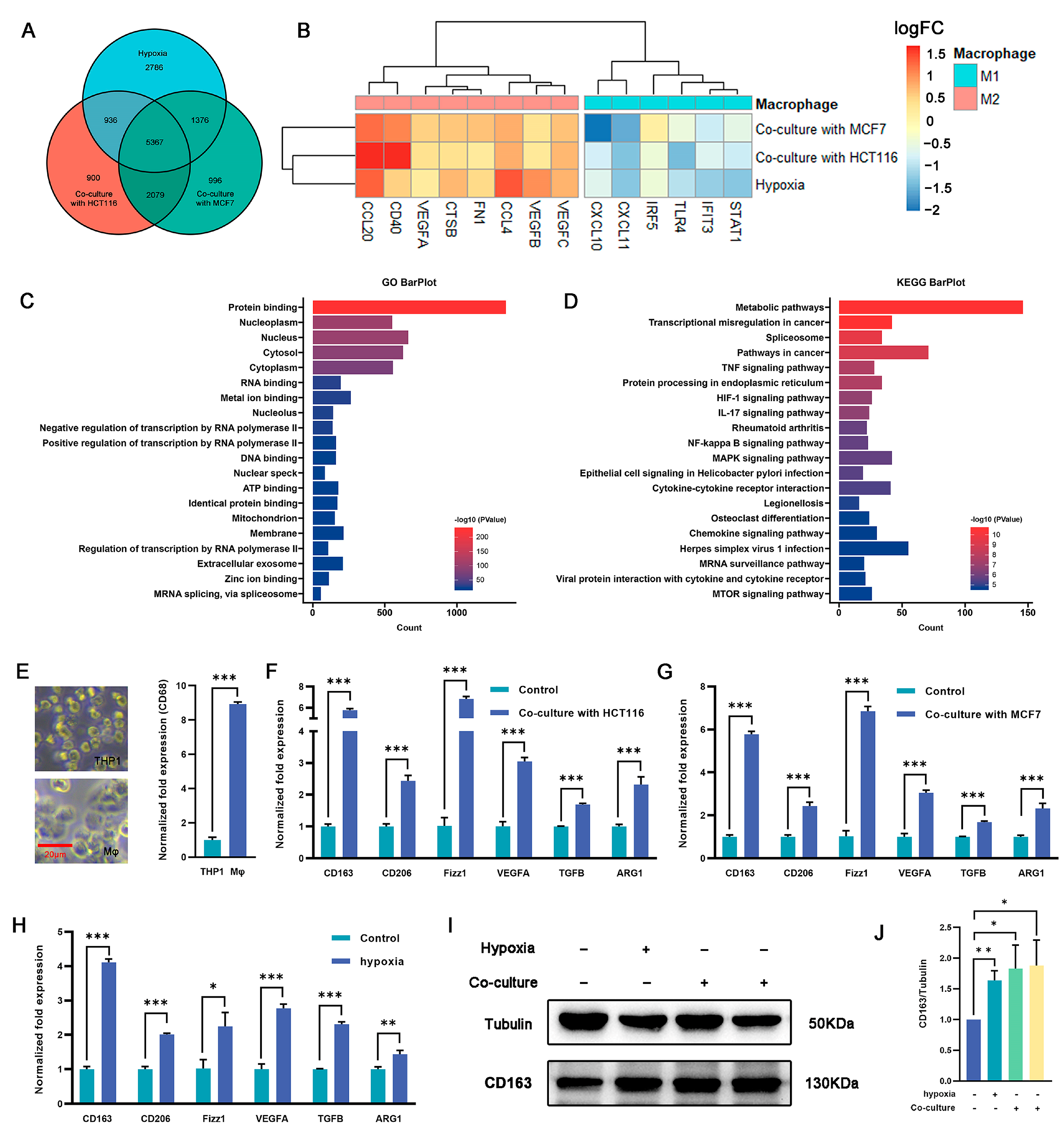
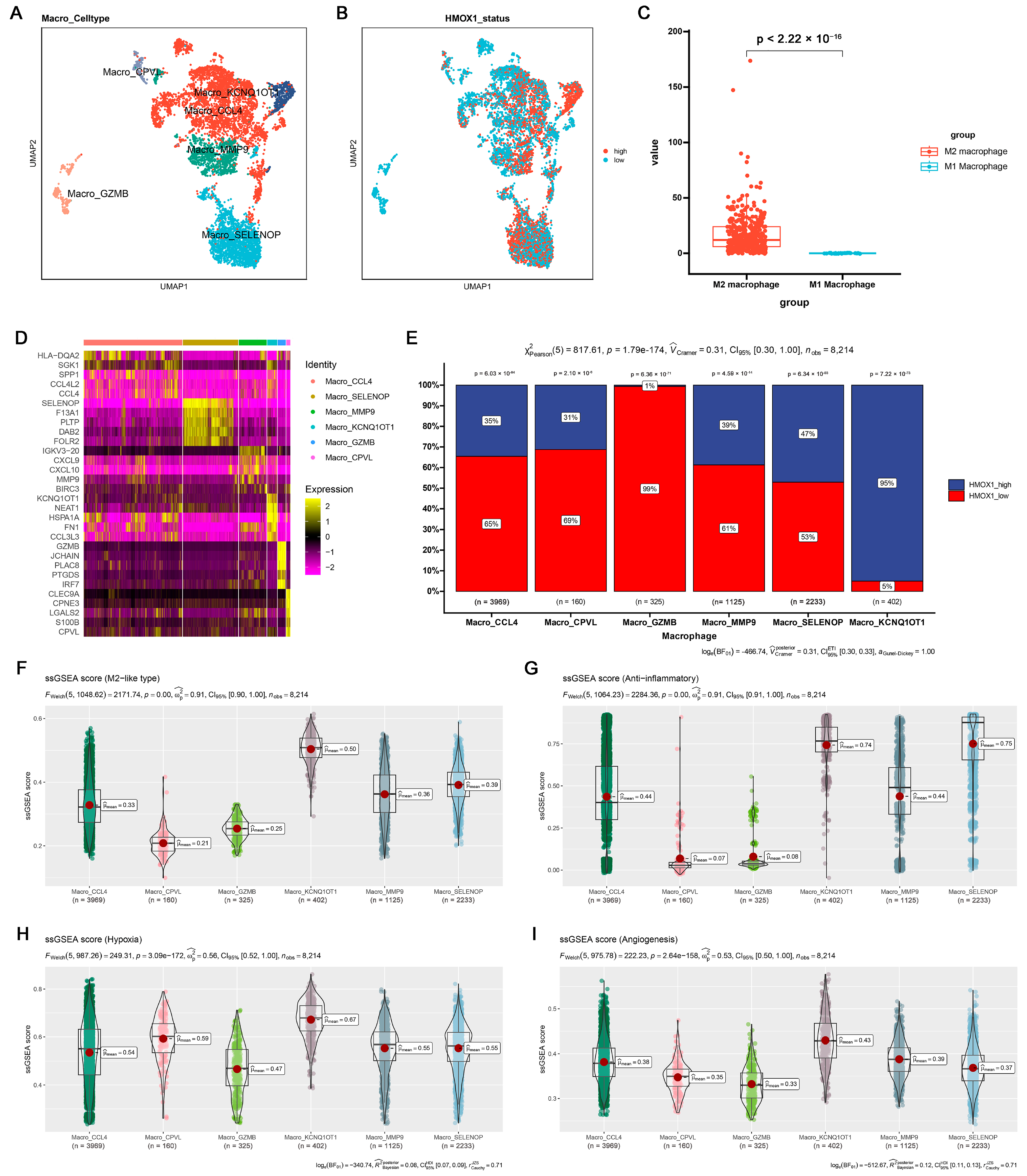
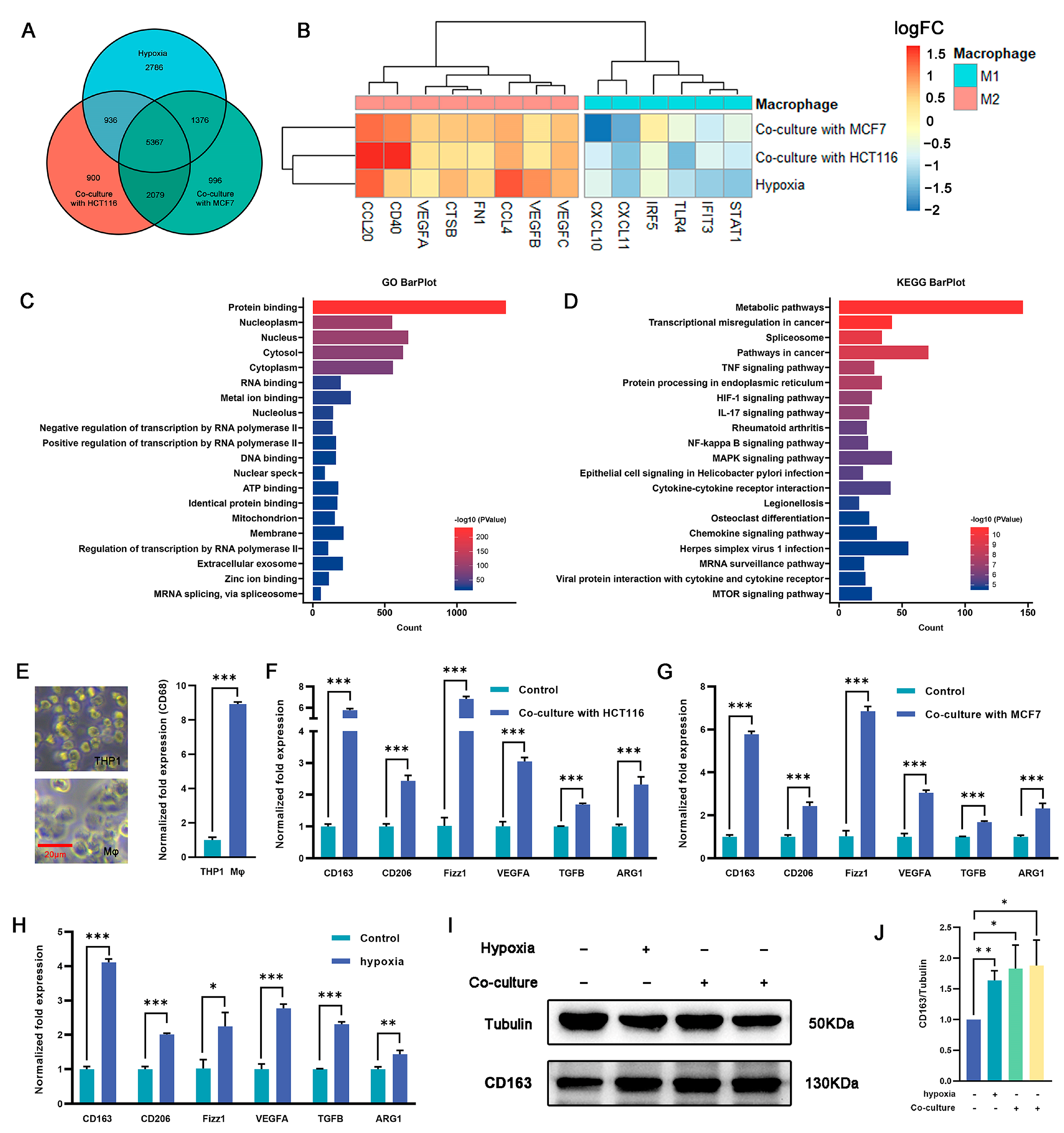
2.2. Induction of M2 Polarization in Macrophages by Coculturing with Cancer Cells and Hypoxia
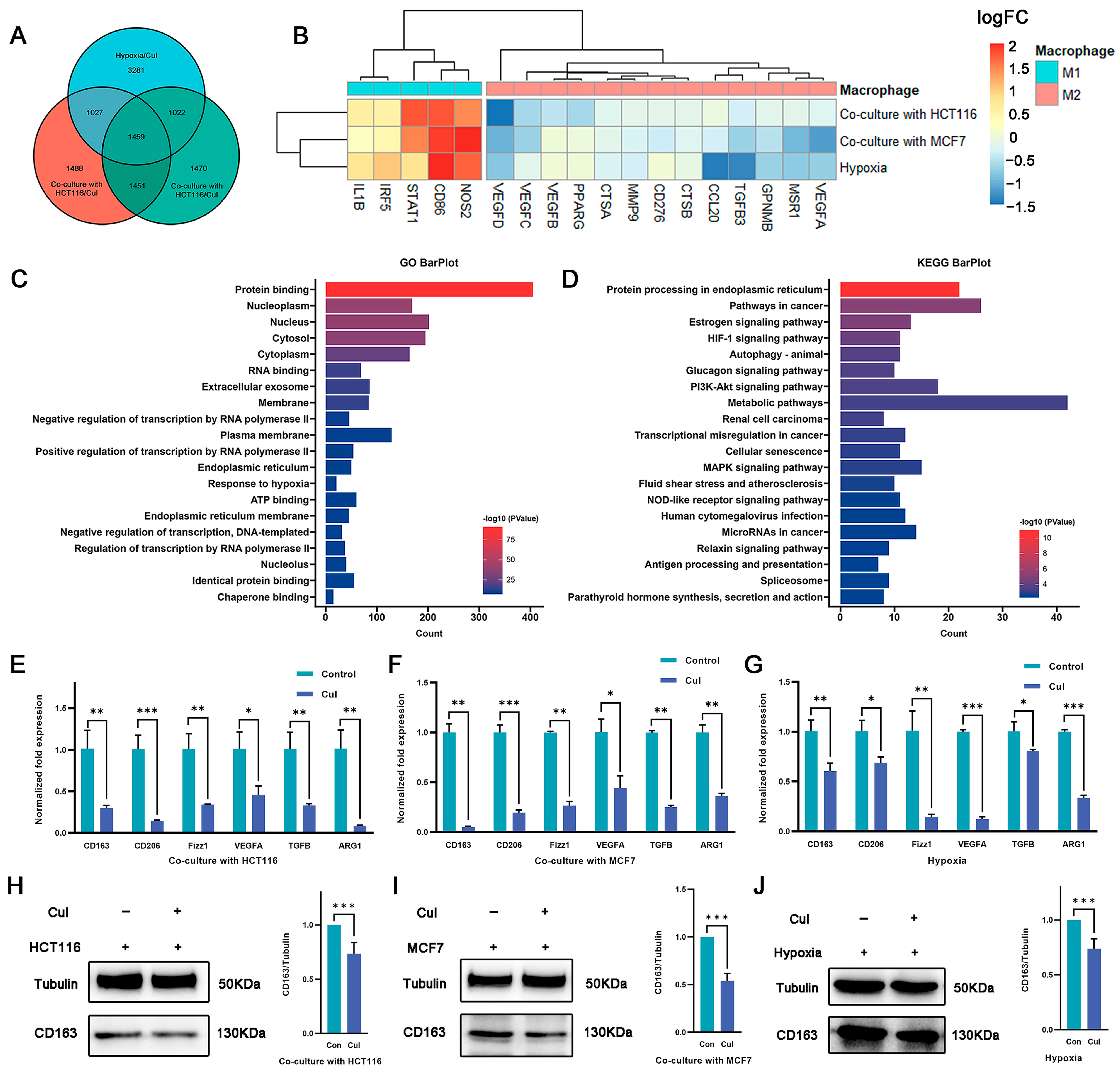
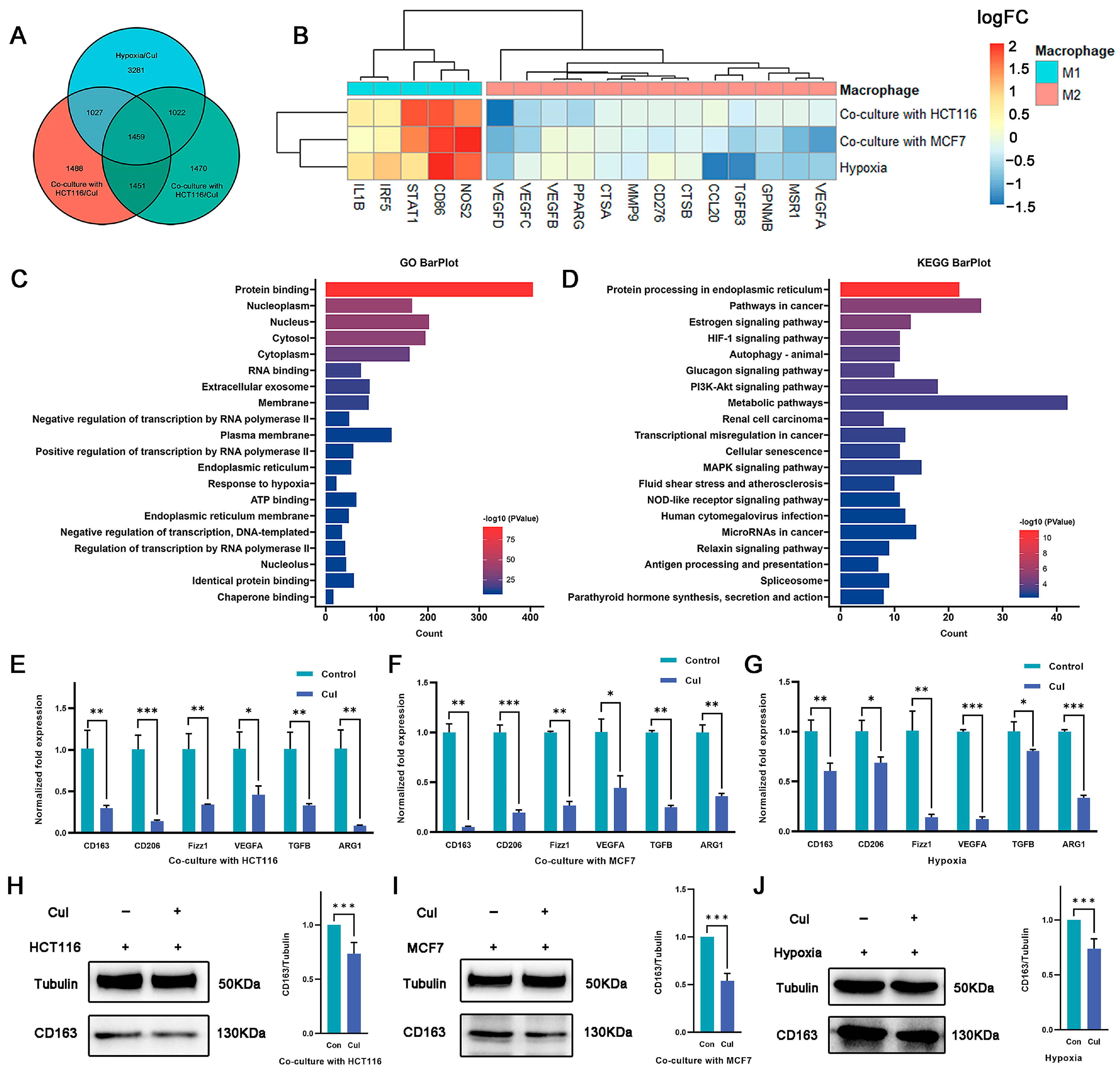
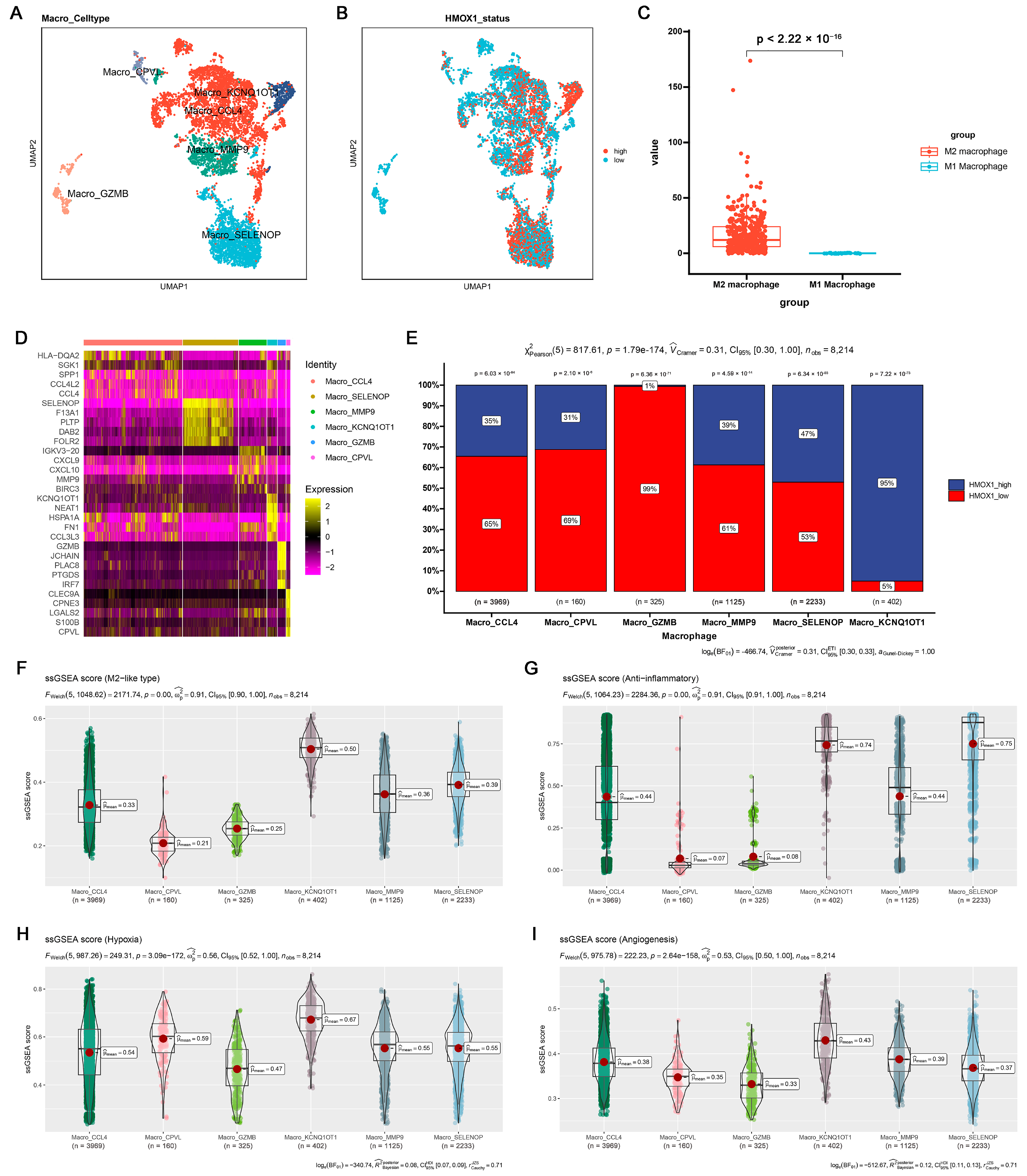
2.3. Cucurbitacin I Inhibits M2-Like Polarization of Macrophages
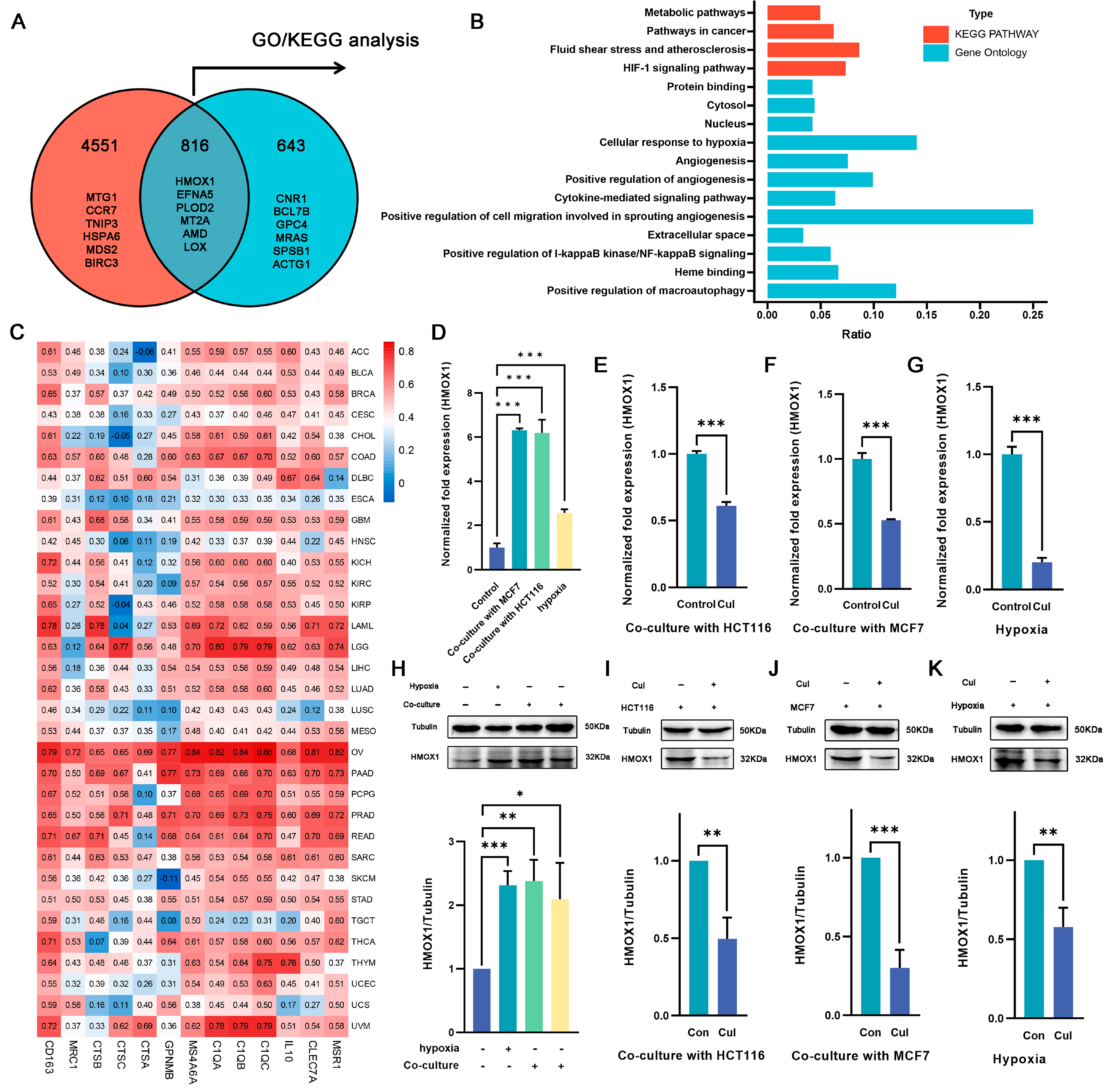
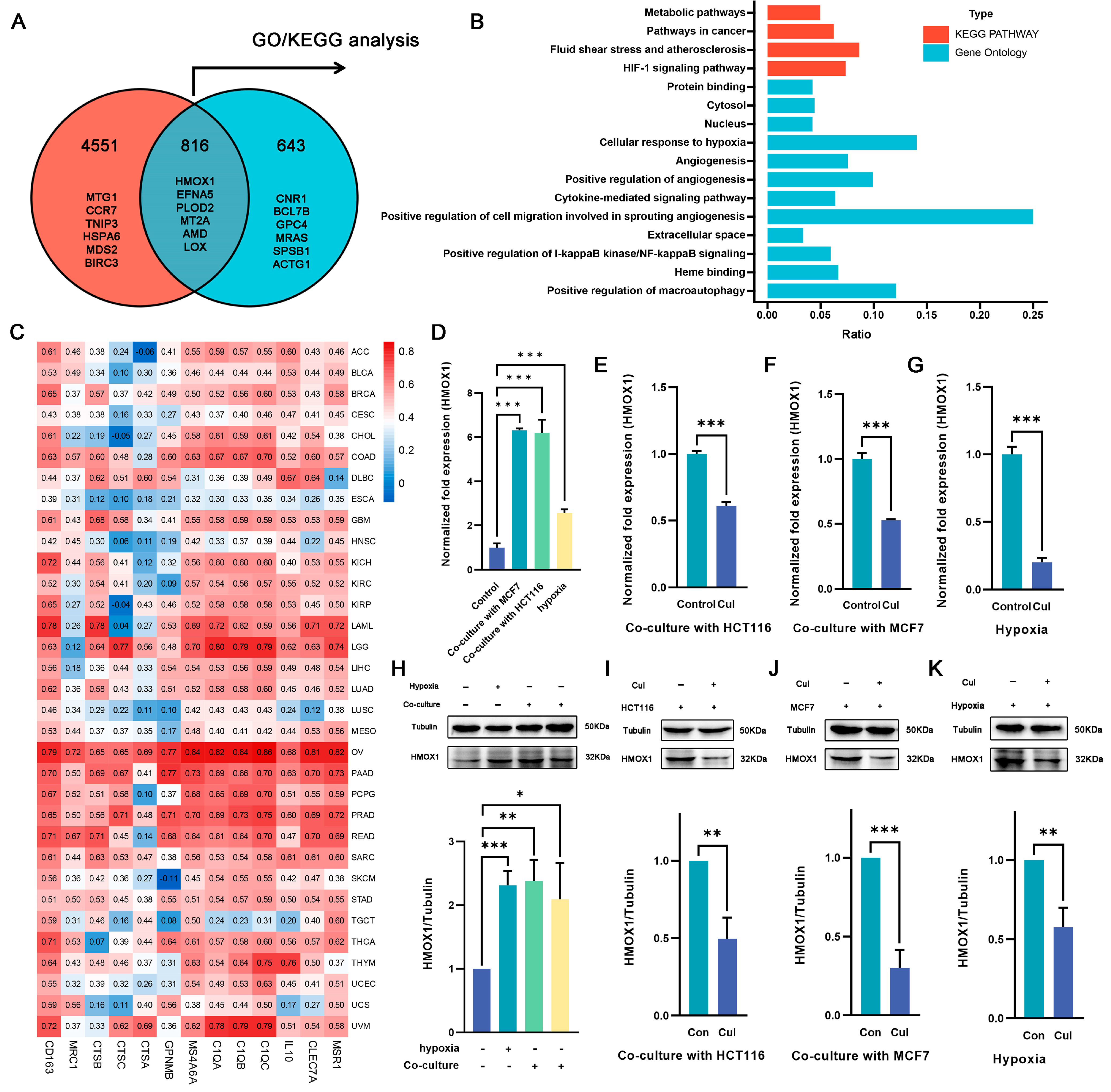
2.4. Hmox1 Was Significantly Downregulated in M2-Like Macrophage under Cucurbitacin I
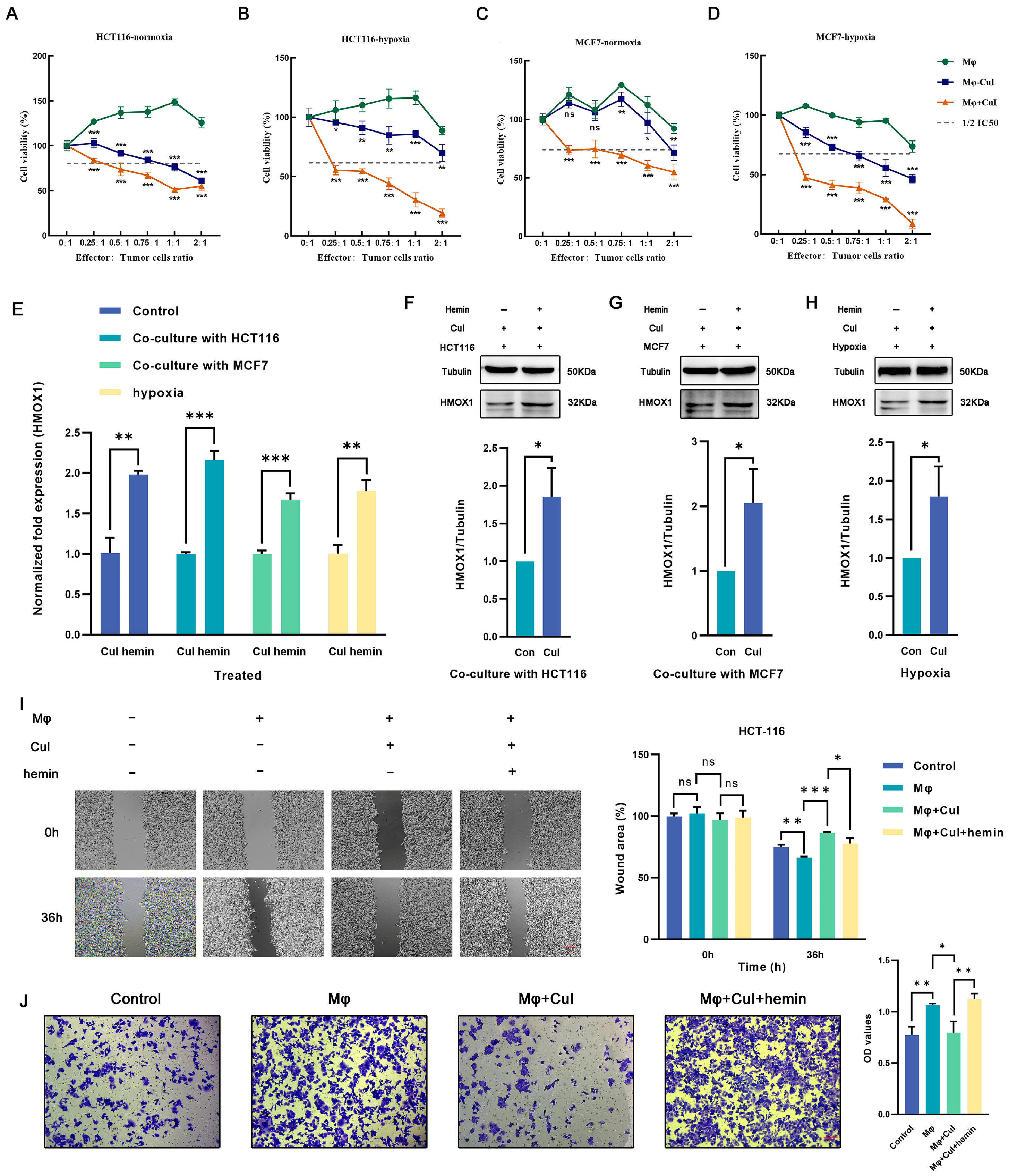
2.5. Hemin Restored the Downregulation of Hmox1 in Macrophages Caused by Cucurbitacin I and the Inhibition of Tumor Cell Migration
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. RNA-seq Analysis
4.3. Differential Expression Analysis
4.4. Functional Enrichment Analysis
4.5. Single-Cell RNA-Sequencing (scRNA-seq) Analysis
4.6. Reagents and Antibodies
4.7. Cell Lines and Cell Culture
4.8. Co-Culture Procedures
4.9. Cell Counting Kit-8 (CCK-8) Assay
4.10. Scratch Wound-Healing Migration Assay
4.11. Colony Formation Assay
4.12. Quantitative Real-Time PCR Analysis (RT-qPCR)
4.13. Western Blotting (WB) Analysis
4.14. Flow Cytometry Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, L.; Li, Z.; Yan, L.; Liu, Y.; Yang, H.; Li, H. Global, regional, and national cancer incidence and death for 29 cancer groups in 2019 and trends analysis of the global cancer burden, 1990–2019. J. Hematol. Oncol. 2021, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Liu, X.; Li, T.; Qiu, W.; Peng, C.; Shen, B.; Zhu, Z. MACC1-AS1 promotes hepatocellular carcinoma cell invasion and proliferation by regulating PAX8. Aging 2020, 12, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, X.; Long, T.; Wang, X.; Zhong, Y.; Ma, Y.; Hu, Z.; Li, Z. FSTL1 interacts with VIM and promotes colorectal cancer metastasis via activating the focal adhesion signalling pathway. Cell Death Dis. 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.J.; Porciani, D.; Burke, D.H. Cancer immunomodulation using bispecific aptamers. Mol. Ther. Nucleic Acids 2022, 27, 894–915. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal 2020, 18, 59. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: Distinct natural history of patients with tumors harboring the T790M mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef]
- Vinogradov, S.; Warren, G.; Wei, X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine 2014, 9, 695–707. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Lim, H.Y.; Weigert, A.; Haussler, A.; Myrczek, T.; Waldner, M.; Labocha, S.; Ferreiros, N.; Geisslinger, G.; Lotsch, J.; et al. Inhibition of GTP cyclohydrolase attenuates tumor growth by reducing angiogenesis and M2-like polarization of tumor associated macrophages. Int. J. Cancer 2013, 132, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.P.; Pinto, M.L.; Pinto, A.T.; Oliveira, M.I.; Pinto, M.T.; Goncalves, R.; Relvas, J.B.; Figueiredo, C.; Seruca, R.; Mantovani, A.; et al. Macrophages stimulate gastric and colorectal cancer invasion through EGFR Y(1086), c-Src, Erk1/2 and Akt phosphorylation and smallGTPase activity. Oncogene 2014, 33, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Mateus-Tique, J.; Mollaoglu, G.; Nielsen, S.R.; Marks, A.; Lakshmi, A.; Khan, J.A.; Wilk, C.M.; Pia, L.; Baccarini, A.; et al. Targeting Macrophages with CAR T Cells Delays Solid Tumor Progression and Enhances Antitumor Immunity. Cancer Immunol. Res. 2022, 10, 1354–1369. [Google Scholar] [CrossRef]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Yang, Z.; Yang, Y.; Peng, Y.; Li, J.; Du, Y.; Sun, Q.; Gao, D.; Yuan, Q.; Zhou, Y.; et al. CRISPR-based in situ engineering tumor cells to reprogram macrophages for effective cancer immunotherapy. Nano Today 2022, 42, 101359. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Ramesh, A.; Kumar, S.; Nandi, D.; Kulkarni, A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, e1904364. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perdigon, M.; Jimaja, S.; Haeni, L.; Bruns, N.; Rothen-Rutishauser, B.; Ruegg, C. Polymersomes-Mediated Delivery of CSF1R Inhibitor to Tumor Associated Macrophages Promotes M2 to M1-Like Macrophage Repolarization. Macromol. Biosci. 2022, 22, e2200168. [Google Scholar] [CrossRef] [PubMed]
- Tanito, K.; Nii, T.; Yokoyama, Y.; Oishi, H.; Shibata, M.; Hijii, S.; Kaneko, R.; Tateishi, C.; Ito, S.; Kishimura, A.; et al. Engineered macrophages acting as a trigger to induce inflammation only in tumor tissues. J. Control Release 2023, 361, 885–895. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Chang, H.W.; Back, J.H.; Lee, S.J.; Park, Y.E.; Kim, E.H.; Hong, Y.; Kwak, G.; Kwon, I.C.; et al. Exosome-guided direct reprogramming of tumor-associated macrophages from protumorigenic to antitumorigenic to fight cancer. Bioact. Mater. 2023, 25, 527–540. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Chong, C.R.; Sullivan, D.J., Jr. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kuang, S.; Song, Q.L.; Liu, M.; Sun, X.X.; Yu, Q. Cucurbitacin I inhibits STAT3, but enhances STAT1 signaling in human cancer cells in vitro through disrupting actin filaments. Acta Pharmacol. Sin. 2018, 39, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Wu, S.; Wang, X.; Zhu, G.; Chen, X.; Ding, Y.; Jiang, W. Cucurbitacin I induces pro-death autophagy in A549 cells via the ERK-mTOR-STAT3 signaling pathway. J. Cell Biochem. 2018, 119, 6104–6112. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhang, B.; Zhang, S.; Duan, C.; Cao, Y.; Kang, W.; Yan, H.; Ding, X.; Zhou, F.; Wu, L.; et al. Low nanomolar concentrations of Cucurbitacin-I induces G2/M phase arrest and apoptosis by perturbing redox homeostasis in gastric cancer cells in vitro and in vivo. Cell Death Dis. 2016, 7, e2106. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xiao, J.; Tang, S.; Lin, X.; Xu, H.; Han, B.; Yang, M.; Liu, F. Cucurbitacin I induces apoptosis in ovarian cancer cells through oxidative stress and the p190B-Rac1 signaling axis. Mol. Med. Rep. 2020, 22, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Spaw, M.; Anant, S.; Thomas, S.M. Stromal contributions to the carcinogenic process. Mol. Carcinog. 2017, 56, 1199–1213. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Lazo, J.S.; Sartorelli, A.C. Classification of antineoplastic agents by their selective toxicities toward oxygenated and hypoxic tumor cells. Cancer Res. 1981, 41, 73–81. [Google Scholar] [PubMed]
- Teicher, B.A.; Holden, S.A.; al-Achi, A.; Herman, T.S. Classification of antineoplastic treatments by their differential toxicity toward putative oxygenated and hypoxic tumor subpopulations in vivo in the FSaIIC murine fibrosarcoma. Cancer Res. 1990, 50, 3339–3344. [Google Scholar]
- Zhu, F.; Li, X.; Chen, S.; Zeng, Q.; Zhao, Y.; Luo, F. Tumor-associated macrophage or chemokine ligand CCL17 positively regulates the tumorigenesis of hepatocellular carcinoma. Med. Oncol. 2016, 33, 17. [Google Scholar] [CrossRef]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q.; et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE(2)/beta-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef]
- She, L.; Qin, Y.; Wang, J.; Liu, C.; Zhu, G.; Li, G.; Wei, M.; Chen, C.; Liu, G.; Zhang, D.; et al. Tumor-associated macrophages derived CCL18 promotes metastasis in squamous cell carcinoma of the head and neck. Cancer Cell Int. 2018, 18, 120. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Long, X.; Zhang, L.; Ye, Y.; Guo, J.; Liu, P.; Zhang, R.; Ning, J.; Yu, W.; Wei, F.; et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology 2018, 7, e1440166. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Xu, D.; Shen, H.; Tian, M.; Chen, W.; Zhang, X. Cucurbitacin I inhibits the proliferation of pancreatic cancer through the JAK2/STAT3 signalling pathway in vivo and in vitro. J. Cancer 2022, 13, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Uremis, N.; Uremis, M.M.; Cigremis, Y.; Tosun, E.; Baysar, A.; Turkoz, Y. Cucurbitacin I exhibits anticancer efficacy through induction of apoptosis and modulation of JAK/STAT3, MAPK/ERK, and AKT/mTOR signaling pathways in HepG2 cell line. J. Food Biochem. 2022, 46, e14333. [Google Scholar] [CrossRef] [PubMed]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Muliaditan, T.; Opzoomer, J.W.; Caron, J.; Okesola, M.; Kosti, P.; Lall, S.; Van Hemelrijck, M.; Dazzi, F.; Tutt, A.; Grigoriadis, A.; et al. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin. Cancer Res. 2018, 24, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Muliaditan, T.; Caron, J.; Okesola, M.; Opzoomer, J.W.; Kosti, P.; Georgouli, M.; Gordon, P.; Lall, S.; Kuzeva, D.M.; Pedro, L.; et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat. Commun. 2018, 9, 2951. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Yuan, G.; Yan, S.F.; Xue, H.; Zhang, P.; Sun, J.T.; Li, G. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 10607–10619. [Google Scholar] [CrossRef]
- Yao, X.; Balamurugan, P.; Arvey, A.; Leslie, C.; Zhang, L. Heme controls the regulation of protein tyrosine kinases Jak2 and Src. Biochem. Biophys. Res. Commun. 2010, 403, 30–35. [Google Scholar] [CrossRef]
- Chen, T.T.; Chen, X.; Zhang, S.S.; Zhu, J.W.; Tang, B.X.; Wang, A.K.; Dong, L.L.; Zhang, Z.W.; Yu, C.X.; Sun, Y.L.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinf. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- Xue, Y.B.; Bao, Y.M.; Zhang, Z.; Zhao, W.M.; Xiao, J.F.; He, S.M.; Zhang, G.Q.; Li, Y.X.; Zhao, G.P.; Chen, R.S.; et al. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 2023, 51, D18–D28. [Google Scholar]
- Fu, S.; Meng, Y.; Zhang, W.; Wang, J.; He, Y.; Huang, L.; Chen, H.; Kuang, J.; Du, H. Transcriptomic Responses of Skeletal Muscle to Acute Exercise in Diabetic Goto-Kakizaki Rats. Front. Physiol. 2019, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Liu, Y.; Liang, K.; Chen, Z.; Ding, K.; Qiu, L.; Wei, J.; Du, H. Cucurbitacin I Reverses Tumor-Associated Macrophage Polarization to Affect Cancer Cell Metastasis. Int. J. Mol. Sci. 2023, 24, 15920. https://doi.org/10.3390/ijms242115920
Gong X, Liu Y, Liang K, Chen Z, Ding K, Qiu L, Wei J, Du H. Cucurbitacin I Reverses Tumor-Associated Macrophage Polarization to Affect Cancer Cell Metastasis. International Journal of Molecular Sciences. 2023; 24(21):15920. https://doi.org/10.3390/ijms242115920
Chicago/Turabian StyleGong, Xiaocheng, Yunfei Liu, Keying Liang, Zixi Chen, Ke Ding, Li Qiu, Jinfen Wei, and Hongli Du. 2023. "Cucurbitacin I Reverses Tumor-Associated Macrophage Polarization to Affect Cancer Cell Metastasis" International Journal of Molecular Sciences 24, no. 21: 15920. https://doi.org/10.3390/ijms242115920
APA StyleGong, X., Liu, Y., Liang, K., Chen, Z., Ding, K., Qiu, L., Wei, J., & Du, H. (2023). Cucurbitacin I Reverses Tumor-Associated Macrophage Polarization to Affect Cancer Cell Metastasis. International Journal of Molecular Sciences, 24(21), 15920. https://doi.org/10.3390/ijms242115920