

Analysis of Potential Biomarkers in Frontal Temporal Dementia: A Bioinformatics Approach
Abstract
:1. Introduction
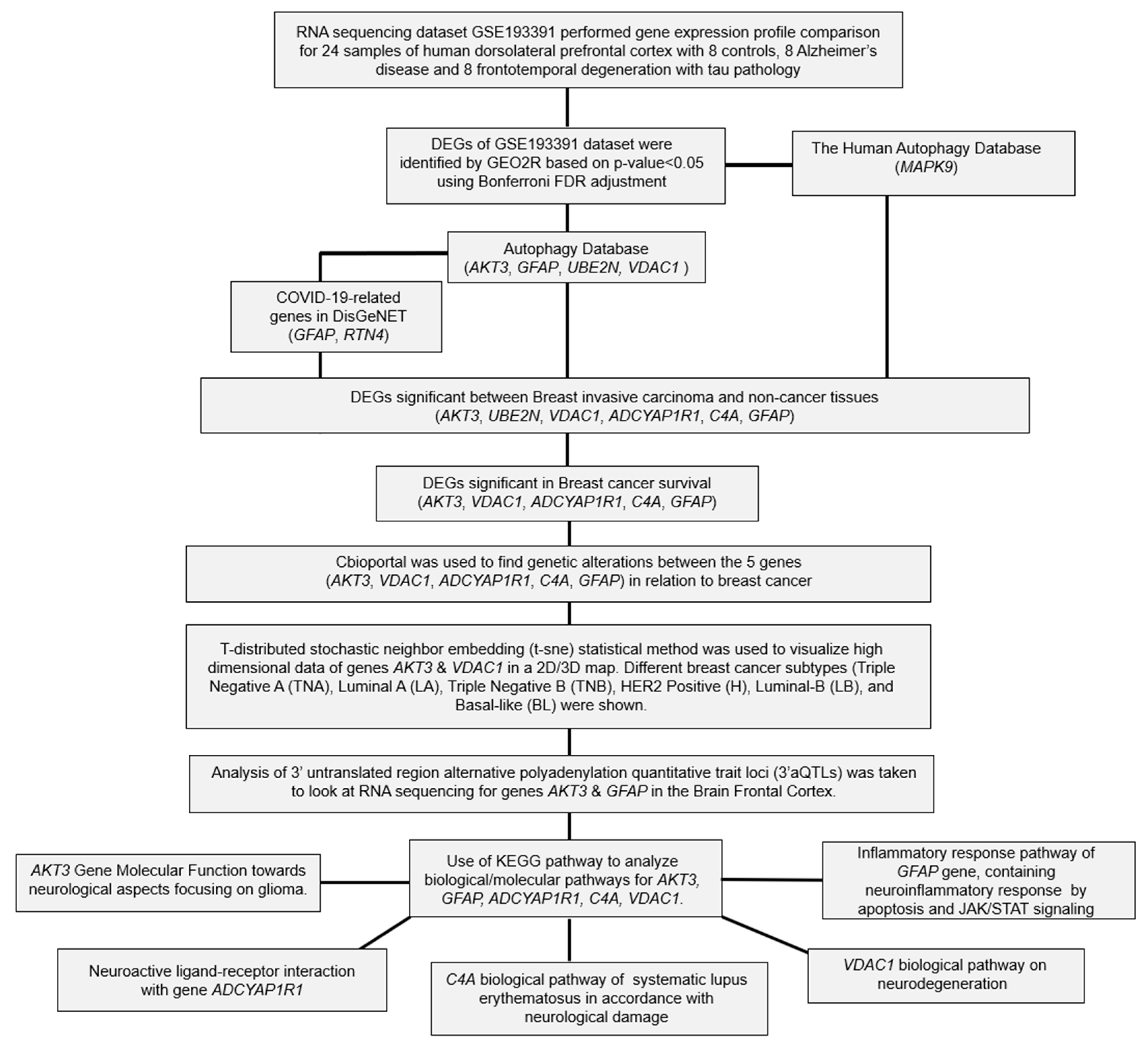
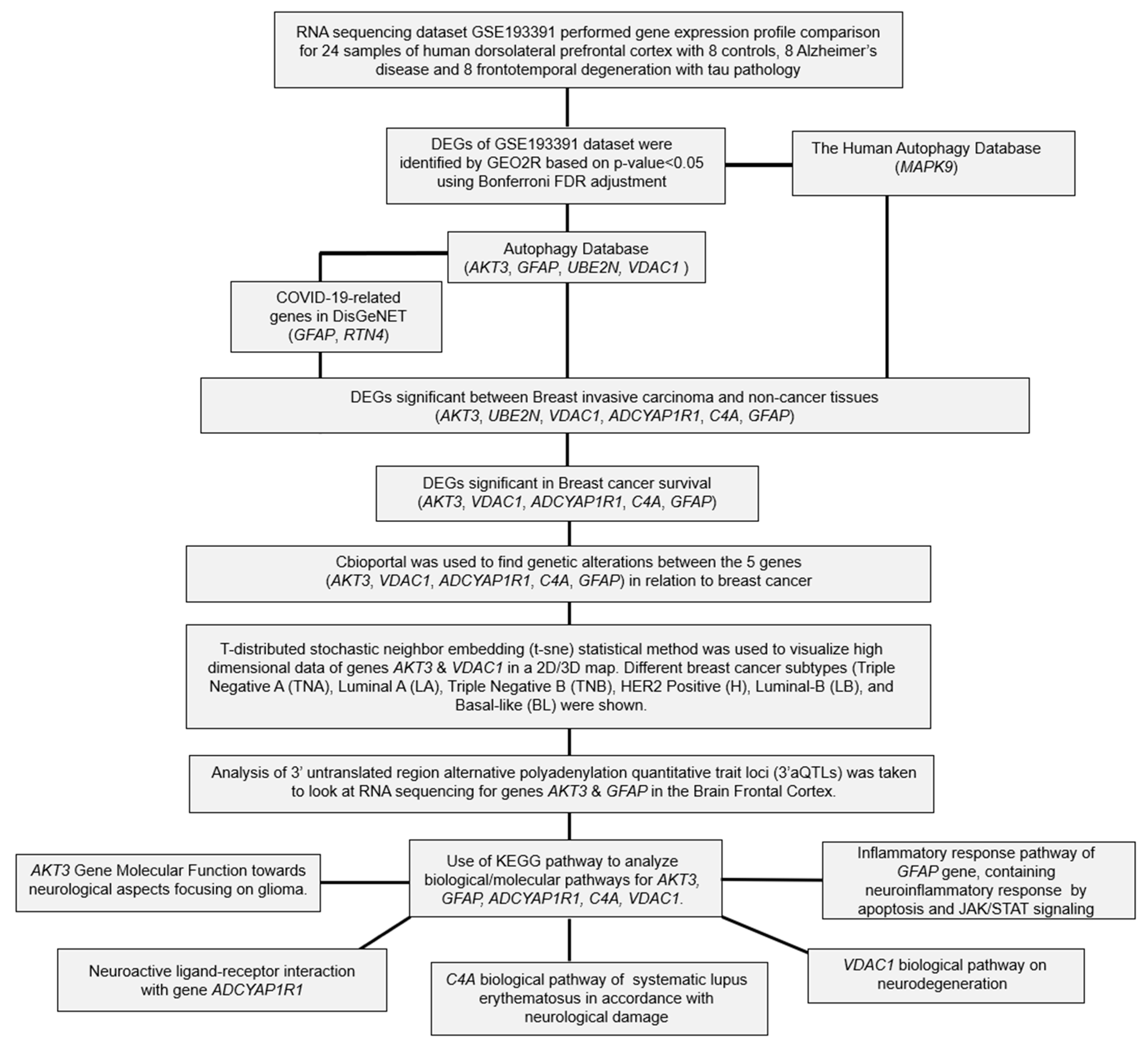
2. Results
2.1. Differential Expression Analysis of FTD Genes
2.2. Analysis of DEGs Involved in Autophagy
2.3. Analysis of the Genes Involved in COVID-19
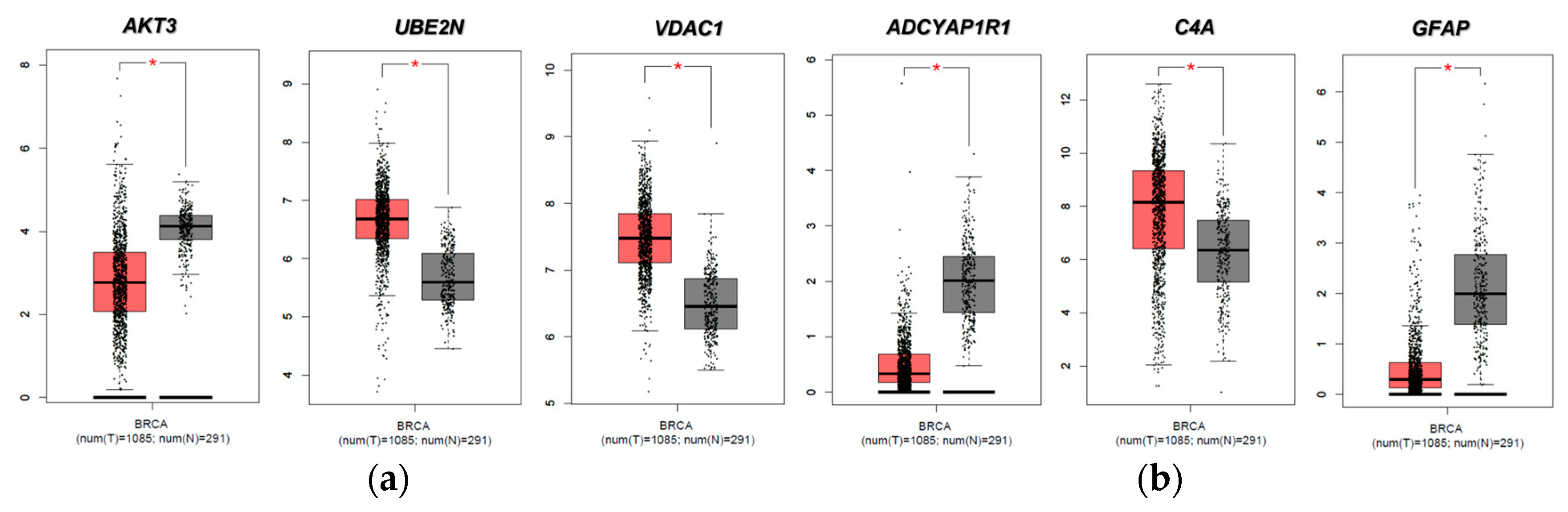
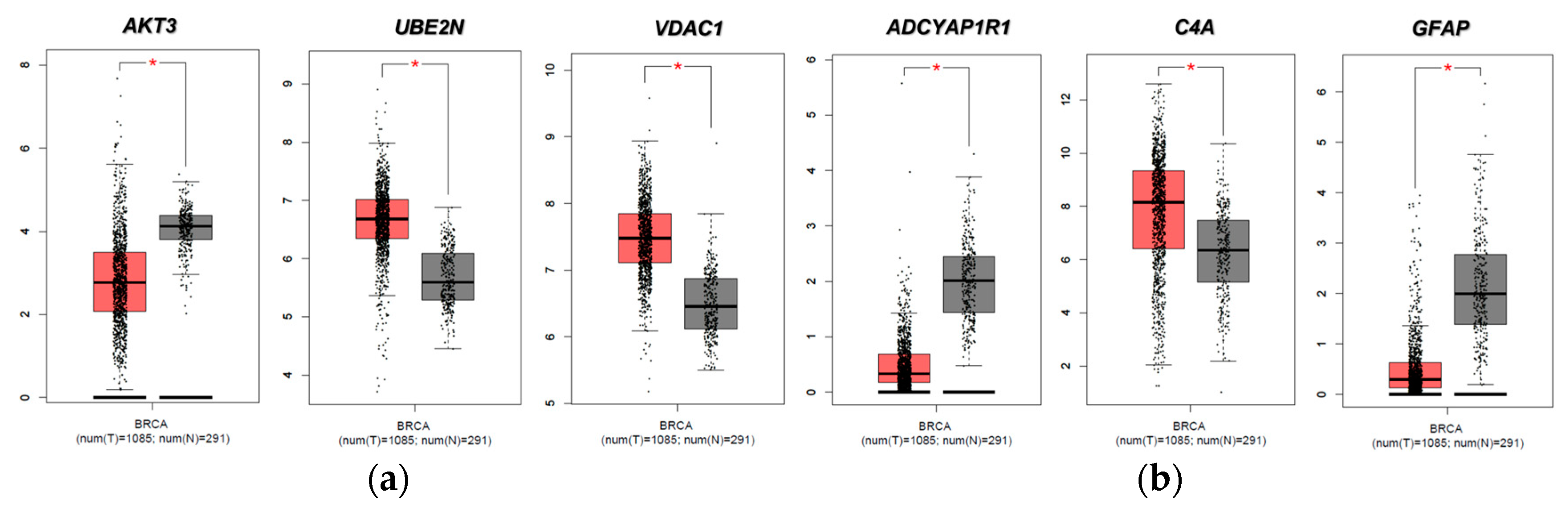
2.4. Evaluation of FTD DEG Expression in Breast Invasive Carcinoma and Survival Analysis
2.5. Pathogenicity Analysis of FTD-Breast Cancer Comorbid Genes
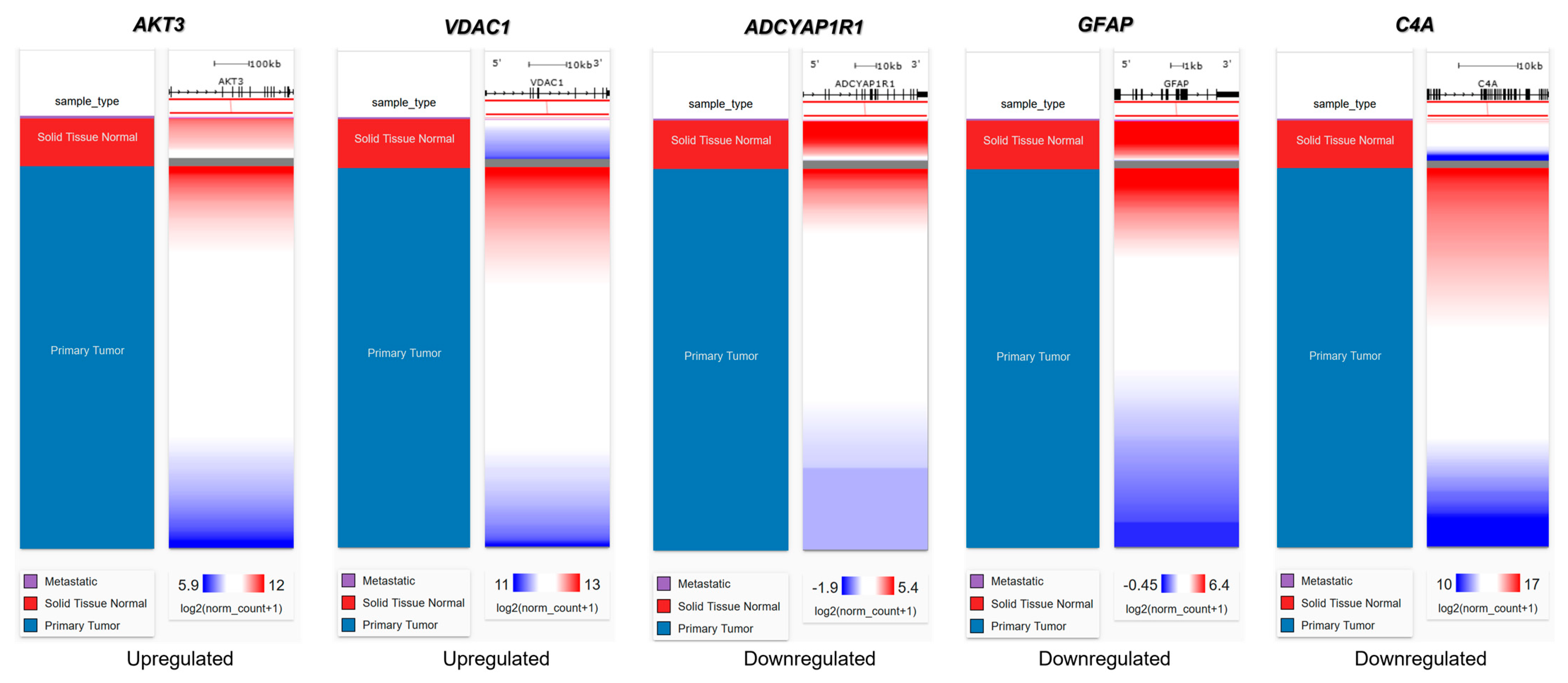
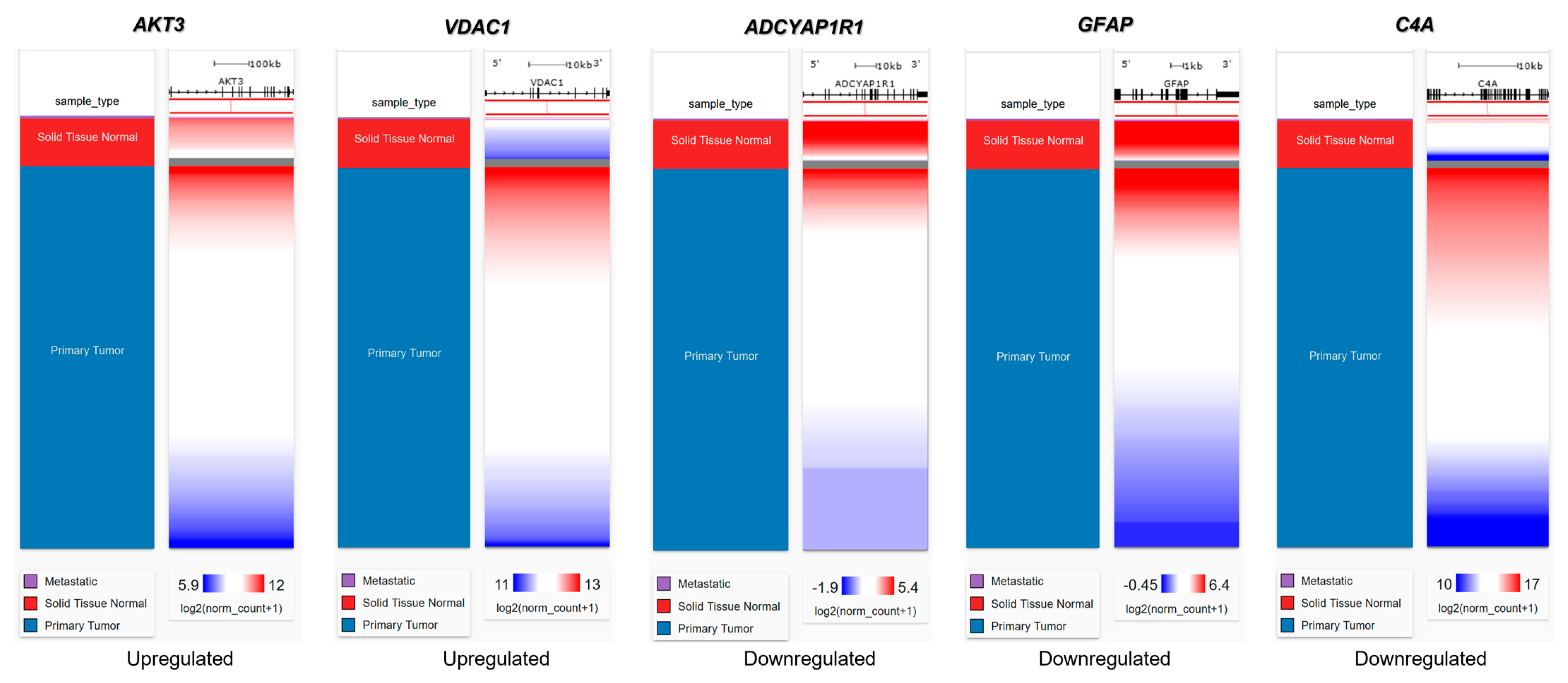
2.6. Genetic Alteration Analysis in FTD-Breast Cancer Comorbid Genes
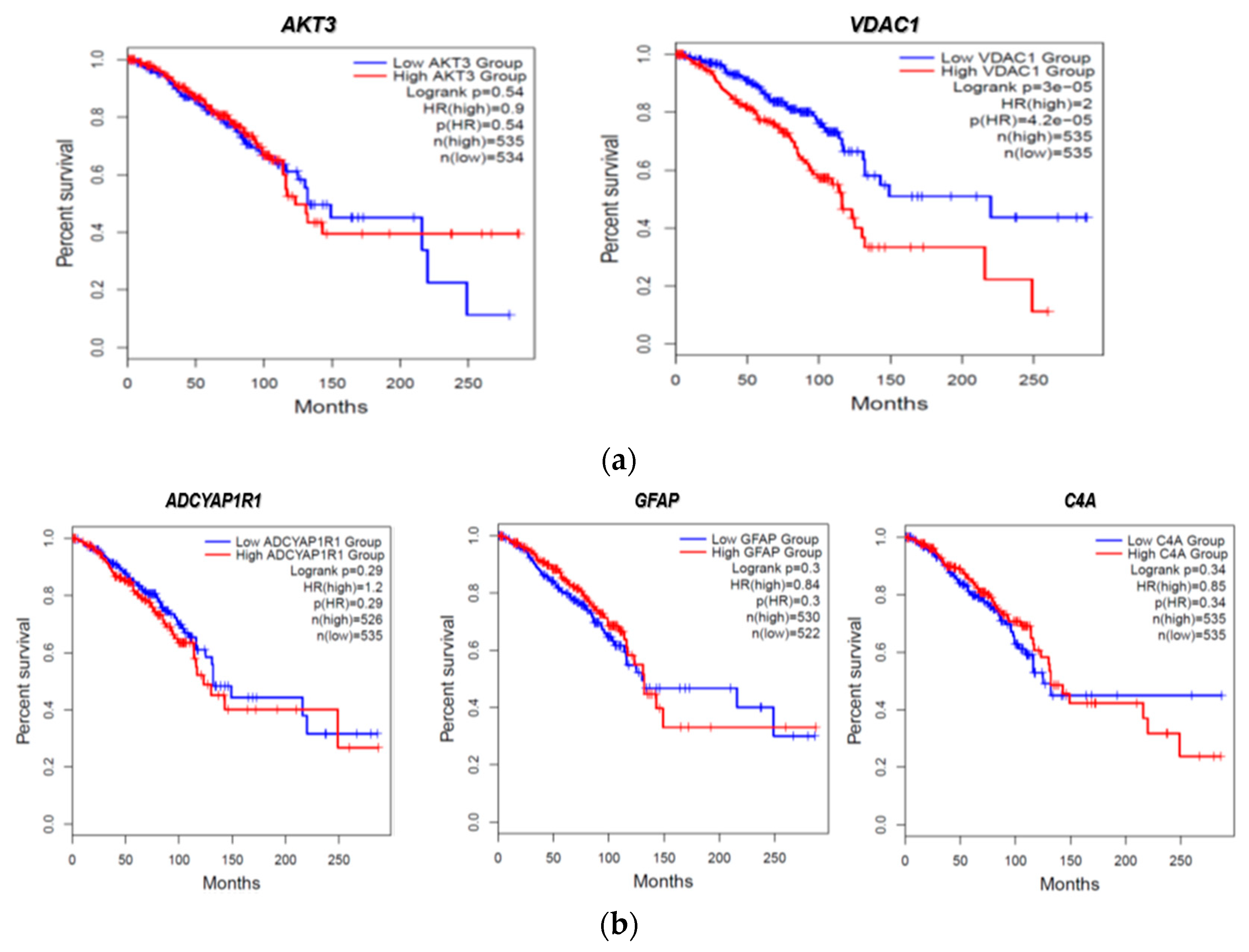
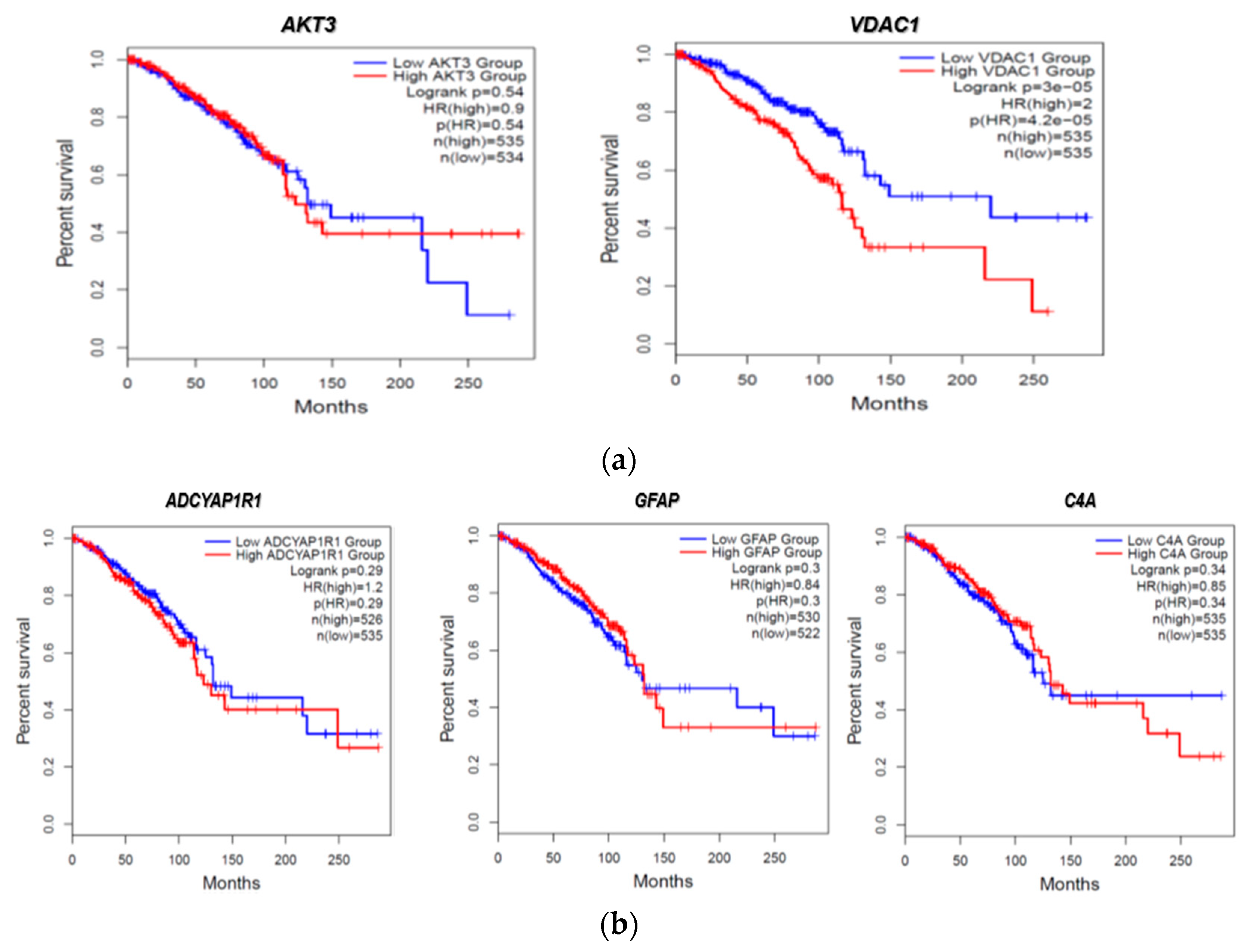
2.7. Clinical Association of FTD-Breast Comorbid Genes
2.8. Pathway Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Data Collection and Identification of Differentially Expressed Genes in FTD
4.2. Identification of Autophagy Genes
4.3. Identification of COVID-19-Associated Genes
4.4. Verification of FTD DEGs in Breast Cancer
4.5. Identification of the Alteration in the DEGs
4.6. Expression Analysis of FTD-Breast Cancer Comorbid Genes
4.7. Identification of RNA Interactions for the FTD-Breast Cancer Comorbid Genes
4.8. Single-Cell Transcriptome Profiling of FTD-Breast Cancer Comorbid Genes
4.9. Prediction of Potential Therapeutic Drugs
4.10. Pathogenicity Gene Score Analysis
4.11. Interpretation of Genetic Variants
4.12. Enrichment of Functional Pathways
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, I.; De Jesus, O. Frontotemporal Lobe Dementia; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Onyike, C.U.; Diehl-Schmid, J. The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 2013, 25, 130–137. [Google Scholar] [CrossRef]
- Guimet, N.M.; Zapata-Restrepo, L.M.; Miller, B.L. Advances in Treatment of Frontotemporal Dementia. J. Neuropsychiatry Clin. Neurosci. 2022, 34, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Zanella, I.; Zacchi, E.; Piva, S.; Filosto, M.; Beligni, G.; Alaverdian, D.; Amitrano, S.; Fava, F.; Baldassarri, M.; Frullanti, E.; et al. C9orf72 Intermediate Repeats Confer Genetic Risk for Severe COVID-19 Pneumonia Independently of Age. Int. J. Mol. Sci. 2021, 22, 6991. [Google Scholar] [CrossRef] [PubMed]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.D.; Warren, J.D.; Fox, N.C.; Rossor, M.N. Presymptomatic studies in genetic frontotemporal dementia. Rev. Neurol. 2013, 169, 820–824. [Google Scholar] [CrossRef]
- Katisko, K.; Haapasalo, A.; Koivisto, A.; Krüger, J.; Hartikainen, P.; Korhonen, V.; Helisalmi, S.; Herukka, S.K.; Remes, A.M.; Solje, E. Low Prevalence of Cancer in Patients with Frontotemporal Lobar Degeneration. J. Alzheimers Dis. 2018, 62, 789–794. [Google Scholar] [CrossRef]
- Bottero, V.; Alrafati, F.; Santiago, J.A.; Potashkin, J.A. Transcriptomic and Network Meta-Analysis of Frontotemporal Dementias. Front. Mol. Neurosci. 2021, 14, 747798. [Google Scholar] [CrossRef]
- Gojobori, T.; Ikeo, K.; Katayama, Y.; Kawabata, T.; Kinjo, A.R.; Kinoshita, K.; Kwon, Y.; Migita, O.; Mizutani, H.; Muraoka, M.; et al. VaProS: A database-integration approach for protein/genome information retrieval. J. Struct. Funct. Genom. 2016, 17, 69–81. [Google Scholar] [CrossRef]
- Cui, Y.; Peng, F.; Wang, D.; Li, Y.; Li, J.S.; Li, L.; Li, W. 3′ aQTL-atlas: An atlas of 3′ UTR alternative polyadenylation quantitative trait loci across human normal tissues. Nucleic Acids Res. 2022, 50, D39–D45. [Google Scholar] [CrossRef]
- Wolfsberg, T.G.; Primakoff, P.; Myles, D.G.; White, J.M. ADAM, a novel family of membrane proteins containing A Disintegrin And Metalloprotease domain: Multipotential functions in cell-cell and cell-matrix interactions. J. Cell Biol. 1995, 131, 275–278. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Finger, E.C. Frontotemporal Dementias. Continuum 2016, 22, 464–489. [Google Scholar] [CrossRef] [PubMed]
- Devenney, E.M.; Ahmed, R.M.; Hodges, J.R. Frontotemporal dementia. Handb. Clin. Neurol. 2019, 167, 279–299. [Google Scholar]
- DeSantis, C.; Siegel, R.; Bandi, P.; Jemal, A. Breast cancer statistics, 2011. CA Cancer J. Clin. 2011, 61, 409–418. [Google Scholar] [CrossRef]
- Costantini, S.; Capone, F.; Polo, A.; Bagnara, P.; Budillon, A. Valosin-Containing Protein (VCP)/p97: A Prognostic Biomarker and Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 10177. [Google Scholar] [CrossRef] [PubMed]
- Aamodt, A.H.; Høgestøl, E.A.; Popperud, T.H.; Holter, J.C.; Dyrhol-Riise, A.M.; Tonby, K.; Stiksrud, B.; Quist-Paulsen, E.; Berge, T.; Barratt-Due, A.; et al. Blood neurofilament light concentration at admittance: A potential prognostic marker in COVID-19. J. Neurol. 2021, 268, 3574–3583. [Google Scholar] [CrossRef]
- Palluzzi, F.; Ferrari, R.; Graziano, F.; Novelli, V.; Rossi, G.; Galimberti, D.; Rainero, I.; Benussi, L.; Nacmias, B.; Bruni, A.C.; et al. A novel network analysis approach reveals DNA damage, oxidative stress and calcium/cAMP homeostasis-associated biomarkers in frontotemporal dementia. PLoS ONE 2017, 12, e0185797. [Google Scholar] [CrossRef]
- Martín-Guerrero, S.M.; Markovinovic, A.; Mórotz, G.M.; Salam, S.; Noble, W.; Miller, C.C.J. Targeting ER-Mitochondria Signaling as a Therapeutic Target for Frontotemporal Dementia and Related Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 2022, 10, 915931. [Google Scholar] [CrossRef]
- Heller, C.; Foiani, M.S.; Moore, K.; Convery, R.; Bocchetta, M.; Neason, M.; Cash, D.M.; Thomas, D.; Greaves, C.V.; Woollacott, I.O.; et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 263–270. [Google Scholar] [CrossRef]
- Reus, L.M.; Pasaniuc, B.; Posthuma, D.; Boltz, T.; Pijnenburg, Y.A.L.; Ophoff, R.A. Gene Expression Imputation Across Multiple Tissue Types Provides Insight Into the Genetic Architecture of Frontotemporal Dementia and Its Clinical Subtypes. Biol. Psychiatry 2021, 89, 825–835. [Google Scholar] [CrossRef]
- Nagatsu, T. The catecholamine system in health and disease -Relation to tyrosine 3-monooxygenase and other catecholamine-synthesizing enzymes. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2007, 82, 388–415. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Johnson, A.G.; Webster, J.A.; Hales, C.M. Glial profiling of human tauopathy brain demonstrates enrichment of astrocytic transcripts in tau-related frontotemporal degeneration. Neurobiol. Aging 2022, 112, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, Y.; Ke, Q.; Wang, Y.; Gong, Z.; Chen, X.; Cai, Y.; Li, S.; Sun, Y.; Peng, X.; et al. ApoE4 associated with severe COVID-19 outcomes via downregulation of ACE2 and imbalanced RAS pathway. J. Transl. Med. 2023, 21, 103. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.; Suzuki, K.; Sugawara, H. The Autophagy Database: An all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res. 2011, 39, D986–D990. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Gambardella, G.; Viscido, G.; Tumaini, B.; Isacchi, A.; Bosotti, R.; di Bernardo, D. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 2022, 13, 1714. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Speir, M.L.; Bhaduri, A.; Markov, N.S.; Moreno, P.; Nowakowski, T.J.; Papatheodorou, I.; Pollen, A.A.; Raney, B.J.; Seninge, L.; Kent, W.J. UCSC cell browser: Visualize your single-cell data. Bioinformatics 2021, 37, 4578–4580. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, Z.; Zhang, W.; Su, G.; Yang, P. Integrated analysis of key pathways and drug targets associated with vogt-Koyanagi-Harada disease. Front. Immunol. 2020, 11, 587443. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Samples | ||||
|---|---|---|---|---|
| Accession | Age | Sex | Race | APOE Status |
| GSM5799484 | 52 | Female | White | E3/4 |
| GSM5799495 | 59 | Male | Black | E2/3 |
| GSM5799498 | 57 | Male | White | E3/3 |
| GSM5799485 | 78 | Female | White | E3/3 |
| GSM5799504 | 56 | Male | White | NA |
| GSM5799489 | 70 | Male | Black | E3/3 |
| GSM5799493 | 75 | Female | White | E3/3 |
| GSM5799506 | 74 | Female | White | E3/3 |
| FTD Samples | ||||
| Accession | Age | Sex | Race | APOE Status |
| GSM5799499 | 81 | Male | White | E3/3 |
| GSM5799500 | 60 | Female | White | E3/4 |
| GSM5799501 | 64 | Male | White | E3/4 |
| GSM5799488 | 71 | Male | White | NA |
| GSM5799505 | 56 | Female | White | NA |
| GSM5799490 | 65 | Female | White | E2/3 |
| GSM5799492 | 65 | Female | White | NA |
| GSM5799494 | 63 | Male | White | E3/3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momin, I.D.; Rigler, J.; Chitrala, K.N. Analysis of Potential Biomarkers in Frontal Temporal Dementia: A Bioinformatics Approach. Int. J. Mol. Sci. 2023, 24, 14910. https://doi.org/10.3390/ijms241914910
Momin ID, Rigler J, Chitrala KN. Analysis of Potential Biomarkers in Frontal Temporal Dementia: A Bioinformatics Approach. International Journal of Molecular Sciences. 2023; 24(19):14910. https://doi.org/10.3390/ijms241914910
Chicago/Turabian StyleMomin, Inara Deedar, Jessica Rigler, and Kumaraswamy Naidu Chitrala. 2023. "Analysis of Potential Biomarkers in Frontal Temporal Dementia: A Bioinformatics Approach" International Journal of Molecular Sciences 24, no. 19: 14910. https://doi.org/10.3390/ijms241914910
APA StyleMomin, I. D., Rigler, J., & Chitrala, K. N. (2023). Analysis of Potential Biomarkers in Frontal Temporal Dementia: A Bioinformatics Approach. International Journal of Molecular Sciences, 24(19), 14910. https://doi.org/10.3390/ijms241914910