
New Insights in Immunometabolism in Neonatal Monocytes and Macrophages in Health and Disease
{kind=link}
{kind=link}
Abstract
:1. Introduction
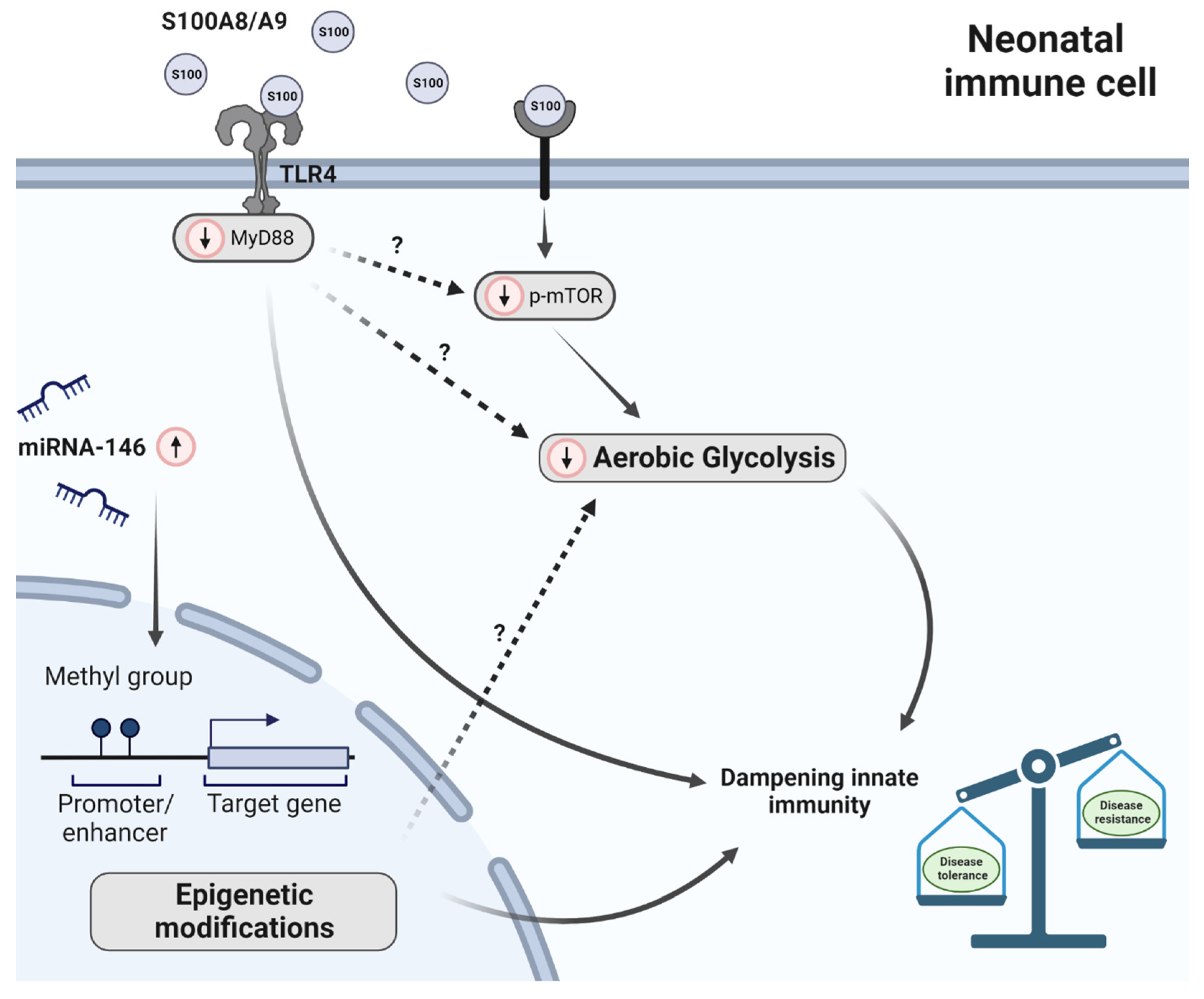
The Neonatal Immune System Is in a Tolerogenic State
2. Carbohydrate Metabolism
2.1. The Glycolytic Metabolic Pathway
2.2. The TCA Cycle and Oxidative Phosphorylation
2.3. Differences in Carbohydrate Metabolism between Adults and Neonates
2.4. External Factors Dictating Immunometabolism in Neonates
3. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kan, B.; Razzaghian, H.R.; Lavoie, P.M. An Immunological Perspective on Neonatal Sepsis. Trends Mol. Med. 2016, 22, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Harbeson, D.; Francis, F.; Bao, W.; Amenyogbe, N.A.; Kollmann, T.R. Energy Demands of Early Life Drive a Disease Tolerant Phenotype and Dictate Outcome in Neonatal Bacterial Sepsis. Front. Immunol. 2018, 9, 1918. [Google Scholar] [CrossRef] [PubMed]
- Hornef, M.W.; Torow, N. ‘Layered immunity’ and the ‘neonatal window of opportunity’–timed succession of non-redundant phases to establish mucosal host–microbial homeostasis after birth. Immunology 2019, 159, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P. Immune-microbe interactions early in life: A determinant of health and disease long term. Science 2022, 376, 945–950. [Google Scholar] [CrossRef]
- Harbeson, D.; Ben-Othman, R.; Amenyogbe, N.; Kollmann, T.R. Outgrowing the Immaturity Myth: The Cost of Defending From Neonatal Infectious Disease. Front. Immunol. 2018, 9, 1077. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef]
- Röszer, T. Metabolic impact of adipose tissue macrophages in the early postnatal life. J. Leukoc. Biol. 2022, 112, 1515–1524. [Google Scholar] [CrossRef]
- World Health Organization. Newborn Mortality; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Singh, M.; Alsaleem, M.; Gray, C.P. Neonatal Sepsis; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sharma, C.M.; Agrawal, R.P.; Sharan, H.; Kumar, B.; Sharma, D.; Bhatia, S.S. “Neonatal Sepsis”: Bacteria & their Susceptibility Pattern towards Antibiotics in Neonatal Intensive Care Unit. J. Clin. Diagn. Res. 2013, 7, 2511–2513. [Google Scholar] [CrossRef]
- Song, W.S.; Park, H.W.; Oh, M.Y.; Jo, J.Y.; Kim, C.Y.; Lee, J.J.; Jung, E.; Lee, B.S.; Kim, K.-S.; Kim, E.A.-R. Neonatal sepsis-causing bacterial pathogens and outcome of trends of their antimicrobial susceptibility a 20-year period at a neonatal intensive care unit. Clin. Exp. Pediatr. 2022, 65, 350–357. [Google Scholar] [CrossRef]
- Kan, B.; Michalski, C.; Fu, H.; Au, H.H.T.; Lee, K.; Marchant, E.A.; Cheng, M.F.; Anderson-Baucum, E.; Aharoni-Simon, M.; Tilley, P.; et al. Cellular metabolism constrains innate immune responses in early human ontogeny. Nat. Commun. 2018, 9, 4822. [Google Scholar] [CrossRef]
- Schlapbach, L.J.; Aebischer, M.; Adams, M.; Natalucci, G.; Bonhoeffer, J.; Latzin, P.; Nelle, M.; Bucher, H.U.; Latal, B. Impact of sepsis on neurodevelopmental outcome in a Swiss national cohort of extremely premature infants. Pediatrics 2011, 128, e348–e357. [Google Scholar] [CrossRef] [PubMed]
- Hentges, C.R.; Silveira, R.C.; Procianoy, R.S.; Carvalho, C.G.; Filipouski, G.R.; Fuentefria, R.N.; Marquezotti, F.; Terrazan, A.C. Association of late-onset neonatal sepsis with late neurodevelopment in the first two years of life of preterm infants with very low birth weight. J. de Pediatr. 2014, 90, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ortgies, T.; Rullmann, M.; Ziegelhöfer, D.; Bläser, A.; Thome, U.H. The role of early-onset-sepsis in the neurodevelopment of very low birth weight infants. BMC Pediatr. 2021, 21, 289. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, I.M. Deconvoluting the ontogeny of hematopoietic stem cells. Cell. Mol. Life Sci. 2014, 71, 957–978. [Google Scholar] [CrossRef]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 10th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.; Chen, W. Modulation of Macrophage Immunometabolism: A New Approach to Fight Infections. Front. Immunol. 2022, 13, 780839. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9, 270. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Human Protein Atlas. 2023. Available online: https://www.proteinatlas.org/ (accessed on 1 March 2023).
- Fu, Y.; Maianu, L.; Melbert, B.R.; Garvey, W. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol. Dis. 2004, 32, 182–190. [Google Scholar] [CrossRef]
- Melkonian, E.A.; Schury, M.P. Biochemistry, Anaerobic Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Williams, N.C.; O’neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Dreschers, S.; Ohl, K.; Lehrke, M.; Möllmann, J.; Denecke, B.; Costa, I.; Vogl, T.; Viemann, D.; Roth, J.; Orlikowsky, T.; et al. Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats. Nat. Commun. 2019, 10, 1685. [Google Scholar] [CrossRef] [PubMed]
- Speer, C.P.; Gahr, M.; Wieland, M.; Eber, S. Phagocytosis-Associated Functions in Neonatal Monocyte-Derived Macrophages. Pediatr. Res. 1988, 24, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Gille, C.; Spring, B.; Tewes, L.; Poets, C.F.; Orlikowsky, T. A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeledEscherichia coli: Comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytom. Part A 2006, 69, 152–154. [Google Scholar] [CrossRef]
- Dreschers, S.; Ohl, K.; Schulte, N.; Tenbrock, K.; Orlikowsky, T.W. Impaired functional capacity of polarised neonatal macrophages. Sci. Rep. 2020, 10, 624. [Google Scholar] [CrossRef]
- Ruland, J.; Hartjes, L. CARD–BCL-10–MALT1 signalling in protective and pathological immunity. Nat. Rev. Immunol. 2019, 19, 118–134. [Google Scholar] [CrossRef]
- Winterberg, T.; Vieten, G.; Meier, T.; Yu, Y.; Busse, M.; Hennig, C.; Hansen, G.; Jacobs, R.; Ure, B.M.; Kuebler, J.F. Distinct phenotypic features of neonatal murine macrophages. Eur. J. Immunol. 2014, 45, 214–224. [Google Scholar] [CrossRef]
- Smith, C.L.; Dickinson, P.; Forster, T.; Craigon, M.; Ross, A.; Khondoker, M.R.; France, R.; Ivens, A.; Lynn, D.J.; Orme, J.; et al. Identification of a human neonatal immune-metabolic network associated with bacterial infection. Nat. Commun. 2014, 5, 4649. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Scicluna, B.P.; Arts, R.J.W.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.L.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Maoldomhnaigh, C.Ó.; Cox, D.J.; Phelan, J.J.; Malone, F.D.; Keane, J.; Basdeo, S.A. The Warburg Effect Occurs Rapidly in Stimulated Human Adult but Not Umbilical Cord Blood Derived Macrophages. Front. Immunol. 2021, 12, 657261. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Domínguez-Andrés, J.; AB Joosten, L.; Netea, M.G. Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 2019, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.-Y.; Arts, R.J.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef]
- Divangahi, M.; Aaby, P.; Khader, S.A.; Barreiro, L.B.; Bekkering, S.; Chavakis, T.; van Crevel, R.; Curtis, N.; DiNardo, A.R.; Dominguez-Andres, J.; et al. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat. Immunol. 2021, 22, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, A.S.; Pirr, S.; Fehlhaber, B.; Mellinger, L.; Burgmann, J.; Busse, M.; Ginzel, M.; Friesenhagen, J.; Köckritz-Blickwede, M.; Ulas, T.; et al. In neonates S100A8/S100A9 alarmins prevent the expansion of a specific inflammatory monocyte population promoting septic shock. FASEB J. 2016, 31, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Willers, M.; Ulas, T.; Völlger, L.; Vogl, T.; Heinemann, A.S.; Pirr, S.; Pagel, J.; Fehlhaber, B.; Halle, O.; Schöning, J.; et al. S100A8 and S100A9 Are Important for Postnatal Development of Gut Microbiota and Immune System in Mice and Infants. Gastroenterology 2020, 159, 2130–2145.e5. [Google Scholar] [CrossRef]
- Ulas, T.; Pirr, S.; Fehlhaber, B.; Bickes, M.S.; Loof, T.G.; Vogl, T.; Mellinger, L.; Heinemann, A.S.; Burgmann, J.; Schöning, J.; et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat. Immunol. 2017, 18, 622–632. [Google Scholar] [CrossRef]
- Henneke, P.; Kierdorf, K.; Hall, L.J.; Sperandio, M.; Hornef, M. Perinatal development of innate immune topology. eLife 2021, 10, e67793. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Ozinsky, A. Toll-like receptors: Key mediators of microbe detection. Curr. Opin. Immunol. 2002, 14, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Levy, O.; Montgomery, R.R.; Goriely, S. Innate Immune Function by Toll-like Receptors: Distinct Responses in Newborns and the Elderly. Immunity 2012, 37, 771–783. [Google Scholar] [CrossRef]
- Viemann, D.; Dubbel, G.; Schleifenbaum, S.; Harms, E.; Sorg, C.; Roth, J. Expression of Toll-Like Receptors in Neonatal Sepsis. Pediatr. Res. 2005, 58, 654–659. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, S.-L.; Li, Y.-P.; Zhang, M.-M. Nucleotide-binding oligomerization domain (NOD) plays an important role in neonatal infection. Int. J. Biol. Macromol. 2018, 121, 686–690. [Google Scholar] [CrossRef]
- Bermick, J.R.; Lambrecht, N.J.; Dendekker, A.D.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M.; Schaller, M.A. Neonatal monocytes exhibit a unique histone modification landscape. Clin. Epigenetics 2016, 8, 99. [Google Scholar] [CrossRef]
- Zentner, G.E.; Tesar, P.J.; Scacheri, P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21, 1273–1283. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Saba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front. Immunol. 2014, 5, 578. [Google Scholar] [CrossRef] [PubMed]
- Lederhuber, H.; Baer, K.; Altiok, I.; Sadeghi, K.; Herkner, K.R.; Kasper, D.C. MicroRNA-146: Tiny Player in Neonatal Innate Immunity? Neonatology 2010, 99, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Katzmarski, N.; Domínguez-Andrés, J.; Cirovic, B.; Renieris, G.; Ciarlo, E.; Le Roy, D.; Lepikhov, K.; Kattler, K.; Gasparoni, G.; Händler, K.; et al. Transmission of trained immunity and heterologous resistance to infections across generations. Nat. Immunol. 2021, 22, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Willis, K.A.; Purvis, J.H.; Myers, E.D.; Aziz, M.M.; Karabayir, I.; Gomes, C.K.; Peters, B.M.; Akbilgic, O.; Talati, A.J.; Pierre, J.F. Fungi form interkingdom microbial communities in the primordial human gut that develop with gestational age. FASEB J. 2019, 33, 12825–12837. [Google Scholar] [CrossRef]
- Kennedy, K.M.; de Goffau, M.C.; Perez-Muñoz, M.E.; Arrieta, M.-C.; Bäckhed, F.; Bork, P.; Braun, T.; Bushman, F.D.; Dore, J.; de Vos, W.M.; et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 2023, 613, 639–649. [Google Scholar] [CrossRef]
- Dietzman, D.E.; Fischer, G.W.; Schoenknecht, F.D. Neonatal escherichia coli septicemia—Bacterial counts in blood. J. Pediatr. 1974, 85, 128–130. [Google Scholar] [CrossRef]
- Zhao, J.; Kim, K.D.; Yang, X.; Auh, S.; Fu, Y.-X.; Tang, H. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7528–7533. [Google Scholar] [CrossRef]
- Ho, P.-C.; Liu, P.-S. Metabolic communication in tumors: A new layer of immunoregulation for immune evasion. J. Immunother. Cancer 2016, 4, 4. [Google Scholar] [CrossRef]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-S.; Chen, Y.-T.; Li, X.; Hsueh, P.-C.; Tzeng, S.-F.; Chen, H.; Shi, P.-Z.; Xie, X.; Parik, S.; Planque, M.; et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat. Immunol. 2023, 24, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Olin, A.; Henckel, E.; Chen, Y.; Lakshmikanth, T.; Pou, C.; Mikes, J.; Gustafsson, A.; Bernhardsson, A.K.; Zhang, C.; Bohlin, K.; et al. Stereotypic Immune System Development in Newborn Children. Cell 2018, 174, 1277–1292.e14. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Pradhan, P.; Braud, L.; Fuchs, H.R.; Gueler, F.; Motterlini, R.; Foresti, R.; Immenschuh, S. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide-A divergent role for glycolysis. Redox Biol. 2019, 22, 101147. [Google Scholar] [CrossRef]
- Verberk, S.G.; de Goede, K.E.; Gorki, F.S.; van Dierendonck, X.A.; Argüello, R.J.; Bossche, J.V.D. An integrated toolbox to profile macrophage immunometabolism. Cell Rep. Methods 2022, 2, 100192. [Google Scholar] [CrossRef]
- Winterberg, T.; Vieten, G.; Feldmann, L.; Yu, Y.; Hansen, G.; Hennig, C.; Ure, B.M.; Kuebler, J.F. Neonatal murine macrophages show enhanced chemotactic capacity upon toll-like receptor stimulation. Pediatr. Surg. Int. 2013, 30, 159–164. [Google Scholar] [CrossRef]
- Goenka, A.; Prise, I.E.; Connolly, E.; Fernandez-Soto, P.; Morgan, D.; Cavet, J.S.; Grainger, J.R.; Nichani, J.; Arkwright, P.D.; Hussell, T. Infant Alveolar Macrophages Are Unable to Effectively Contain Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 486. [Google Scholar] [CrossRef]
- Woods, P.S.; Kimmig, L.M.; Meliton, A.Y.; Sun, K.A.; Tian, Y.; O’leary, E.M.; Gökalp, G.A.; Hamanaka, R.B.; Mutlu, G.M. Tissue-Resident Alveolar Macrophages Do Not Rely on Glycolysis for LPS-induced Inflammation. Am. J. Respir. Cell Mol. Biol. 2020, 62, 243–255. [Google Scholar] [CrossRef]
- Pereverzeva, L.; van Linge, C.C.; Schuurman, A.R.; Klarenbeek, A.M.; Moral, I.R.; Otto, N.A.; Peters-Sengers, H.; Butler, J.M.; Schomakers, B.V.; van Weeghel, M.; et al. Human alveolar macrophages do not rely on glucose metabolism upon activation by lipopolysaccharide. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166488. [Google Scholar] [CrossRef]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef]
- Skibsted, S.; Bhasin, M.K.; Aird, W.C.; Shapiro, N.I. Bench-to-bedside review: Future novel diagnostics for sepsis-a systems biology approach. Crit. Care 2013, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Fanos, V.; Caboni, P.; Corsello, G.; Stronati, M.; Gazzolo, D.; Noto, A.; Lussu, M.; Dessì, A.; Giuffrè, M.; Lacerenza, S.; et al. Urinary 1H-NMR and GC-MS metabolomics predicts early and late onset neonatal sepsis. Early Hum. Dev. 2014, 90, S78–S83. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, K.; Chatziioannou, A.C.; Thomaidou, A.; Gika, H.; Mikros, E.; Benaki, D.; Diamanti, E.; Agakidis, C.; Raikos, N.; Drossou, V.; et al. Urine metabolomics in neonates with late-onset sepsis in a case-control study. Sci. Rep. 2017, 7, 45506. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jong, R.; Tenbrock, K.; Ohl, K. New Insights in Immunometabolism in Neonatal Monocytes and Macrophages in Health and Disease. Int. J. Mol. Sci. 2023, 24, 14173. https://doi.org/10.3390/ijms241814173
de Jong R, Tenbrock K, Ohl K. New Insights in Immunometabolism in Neonatal Monocytes and Macrophages in Health and Disease. International Journal of Molecular Sciences. 2023; 24(18):14173. https://doi.org/10.3390/ijms241814173
Chicago/Turabian Stylede Jong, Renske, Klaus Tenbrock, and Kim Ohl. 2023. "New Insights in Immunometabolism in Neonatal Monocytes and Macrophages in Health and Disease" International Journal of Molecular Sciences 24, no. 18: 14173. https://doi.org/10.3390/ijms241814173
APA Stylede Jong, R., Tenbrock, K., & Ohl, K. (2023). New Insights in Immunometabolism in Neonatal Monocytes and Macrophages in Health and Disease. International Journal of Molecular Sciences, 24(18), 14173. https://doi.org/10.3390/ijms241814173