

Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease


Abstract
1. Introduction
2. Cyclic Nucleotide-Dependent Kinase Regulation of mTOR Signaling
2.1. Regulation of mTOR Signaling by PKA
2.2. Regulation of mTOR Signaling by PKG
2.3. Indirect Regulation of mTOR Signaling

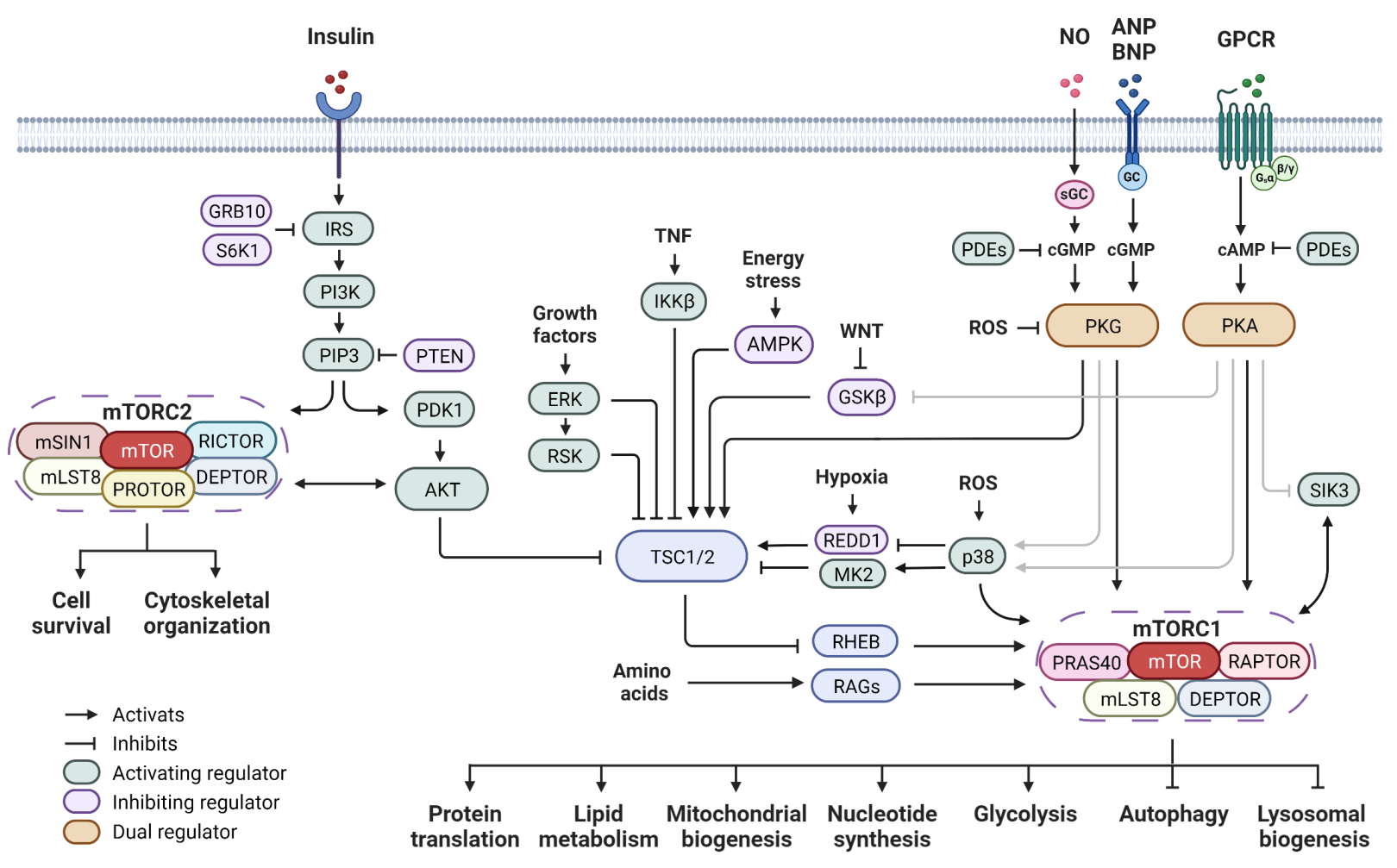{kind=link}
| No. | Reagents | Signaling Cascade and Effects | Mechanism | Targets | Readouts | Ref. |
|---|---|---|---|---|---|---|
| Inhibition by cAMP | ||||||
| 1 | Forskolin, IBMX | Inhibits interleukin-2-dependent mTORC1 activation in IL-2-responsive lymphoid cells | Activates PKA and inhibits PI3K and S6K1 activation | PI3K | p-S6K1 | [23] |
| 2 | Forskolin, cAMP analogue | Prevents the effect of insulin on mTORC1 activation in 3T3-L1 adipocyte | Inhibits mTOR phosphorylation and activation | mTOR | p-4E-BP | [24] |
| 3 | Forskolin, IBMX | Inhibits insulin and amino acid-stimulated mTORC1 activation in MEFs and HEK293 cells | Promotes mTOR complex disassembly and inhibits mTOR catalytic activity | mTOR | p-S6K1, p-4E-BP | [25] |
| 4 | Forskolin, IBMX | Inhibits serum-stimulated mTORC2 activation after prolonged cAMP elevation in MEFs and HEK293 cells | Promotes mTOR complex disassembly and inhibits mTOR catalytic activity | mTOR | p-PKB, p-PKC | [25] |
| 5 | Forskolin, cAMP analogue | Inhibits mTORC1 activation in Ret/PTC1-positive thyroid carcinoma cell TPC-1 | Inhibits the Raf/ERK cascade and depends on PKA | Raf/ERK cascade | p-S6K1, p-4E-BP | [26] |
| 6 | cAMP analogue | Inhibits EGF-stimulated mTOR activity in Swiss 3T3, HEK293, COS, and Rat2 cells | Inhibits PI3K activity, PDK1 translocation to plasma membrane, and AKT phosphorylation | PI3K/PDK/AKT cascade | p-S6K1 | [27] |
| 7 | Glucagon, cAMP analogue | Inhibits insulin- and amino acid-induced mTORC1 activation in serum-free primary rat hepatocytes | NA | NA | p-S6K1, p-4E-BP1 | [28] |
| 8 | Glucagon | Represses both basal and amino acid-induced mTORC1 activation in perfused rat liver | Activates PKA and enhances phosphorylation of LKB1 Thr172 and AMPK Ser428 | LKB1/AMPK pathway | p-S6K1, p-4E-BP1 | [29] |
| 9 | Glucagon | Acts in a dominant manner to repress insulin-induced mTORC1 signaling in perfused rat liver | Reduces assembly of the mTORC1-eIF3 complex | mTORC1-eIF3 complex | p-S6K1, p-4E-BP1 | [30] |
| 10 | PTH/PTHrP | Leads to mTORC1 inhibition during skeletal development | Inhibits SIK3 and prevents DEPTOR degradation | DEPTOR | p-S6K, p-S6 | [31] |
| Activation by cAMP | ||||||
| 11 | Forskolin, GLP-1RA exenatide | Activates mTORC1 in isolated rat islets | Mobilizes intracellular Ca2+ influx and upregulates ATP production | KATP channel | p-S6K1 | [35] |
| 12 | Adrenalin, cAMP analogue | Potentiates insulin-stimulated mTORC1 activation in soleus muscles ex vivo | Depends on cAMP and Epac but not PKA | EPAC | p-S6K1 | [36] |
| 13 | GLP-1, Forskolin | Leads to S6 phosphorylation in the pancreatic beta cell line MIN6 and islets | Depends on PKA phosphorylation of S6 at Ser235/Ser236 | S6 | p-S6 | [37] |
| 14 | TSH | Activates mTORC1 in PC Cl3 rat thyroid cells | Acts in part via phosphorylation of PRAS40 Thr246 by PKA and augments 4E-BP1 binding to RAPTOR | PRAS40 | p-S6K1, p-4E-BP1 | [38] |
| 15 | hCG, Forskolin | Activates mTORC1 in theca-interstitial cells | Involved in cAMP-dependent activation of AKT | AKT | p-S6K, p-S6, p-4E-BP1 | [39] |
| 16 | cAMP | Leads mTORC1 activation in HEK293 cells | Disrupts the interaction of PDE4D with RHEB and increases the interaction between RHEB and mTOR | PDE4D and RHEB | p-S6K1, p-4EBP1 | [40] |
| 17 | β-AR agonist | Leads to mTORC1 activation in adipocyte and mouse adipose tissue | Direct PKA phosphorylation of RAPTOR Ser791 and mTOR Ser1276, Ser1288, and Ser2112 | RAPTOR and mTOR | p-S6K1 | [41] |
| 18 | GLP-1RA liraglutide | Promotes mTORC1 activation in CHO cells expressing GLP-1R | Direct PKA phosphorylation of RAPTOR Ser791 | RAPTOR | p-S6K1 | [62] |
| Activation by cGMP | ||||||
| 19 | Natriuretic peptides | Leads to mTORC1 activation in adipocyte and mouse adipose tissue | Direct PKG phosphorylation of RAPTOR Ser791 | RAPTOR | p-S6K, p-S6 | [42] |
| Inhibition by cGMP | ||||||
| 20 | PKG | Inhibits mTORC1 and promotes autophagy activity | Direct PKG1 phosphorylation of TSC2 Ser1365 and Ser1366, PKG1α C42 oxidation reduces TSC2 phosphorylation and amplifies mTORC1 activity | TSC2 | p-S6K1, p-4E-BP1 | [44,46] |
| Indirect Regulation via p38 | ||||||
| 21 | p38 | Activates mTORC1 by inhibiting TSC2 | Controls TSC2 Ser1210 phosphorylation through MAPKAPK2 (MK2) and increases 14-3-3 binding | TSC2 | p-TSC2 | [49] |
| 22 | p38β | Leads to TORC1 activation in stressed mammalian and Drosophila tissue culture | Occurs downstream of or in parallel to TSC2 and upstream of Rag activation | ND | p-S6K1, p-4E-BP1 | [50] |
| 23 | p38 | Leads to mTORC1 activation in reperfused heart and reoxygenated or H2O2-treated cardiomyocytes (NRVMs) | ROS downregulates REDD1 and promotes TSC2/14-3-3 association | TSC2 | p-S6 | [51] |
| 24 | p38γ/δ | Leads to mTORC1 activation in postnatal cardiac hypertrophic growth | Phosphorylates DEPTOR and leads to its degradation and mTOR activation | DEPTOR | p-mTOR, p-S6K1, p-S6 | [52] |
| Indirect Regulation via GSK3β | ||||||
| 25 | GSK3β | Phosphorylates TSC2 and results in mTORC1 inhibition | PKA physically associates with, phosphorylates, and inactivates both isoforms of GSK3 | TSC2 | p-AKT | [57,58] |
| 26 | GSK3β | Required for mTORC2 inhibition during ER stress | Phosphorylates RICTOR at Ser1235, interferes with the binding of AKT to mTORC2 and thus inhibits AKT | RICTOR | p-AKT | [59] |
| 27 | GSK3β | Required for mTORC2 inhibition | Directly phosphorylates RICTOR at Thr1695, recruits FBW7 and leads to RICTOR degradation | RICTOR | p-AKT, p-SGK1, p-PKCα | [60] |
| 28 | GSK3β | Required for mTORC2 activation in response to BDNF or insulin stimulation in cultured neurons and mouse brain | Increases prosurvival signaling | NA | p-S6, p-AKT | [61] |
3. Role of mTOR Signaling in Cardiometabolic Disease
3.1. Role of mTOR Signaling in Obesity
3.2. Role of mTOR Signaling in Fatty Liver Disease
3.3. Role of mTOR Signaling in Cardiac Remodeling
| No. | Genes | Tissues | Models | Phenotypes | Ref. |
|---|---|---|---|---|---|
| Adipose tissue | |||||
| 1 | Raptor | Adipocyte Fabp4/aP2-Cre | HFD | Substantially less adipose tissue, protected against diet-induced obesity, hypercholesterolemia, and insulin resistance | [74] |
| 2 | Raptor | Adipocyte Adipoq-Cre | HFD | Progressive lipodystrophy with hepatic steatosis and insulin intolerance, resistant to diet-induced obesity, severe hepatomegaly | [75] |
| 3 | Raptor | Adipocyte Adipoq-Cre | HFD | Fails to completely suppress lipolysis in the fed state and displays prominent hypertriglyceridemia and hypercholesterolemia | [79] |
| 4 | Raptor | Adipocyte Adipoq-Cre | Cold, βAR agonist | Refractory to the β-adrenergic stimulation of UCP1 expression and expansion of beige/brite adipocytes in WAT | [41] |
| 5 | Raptor | Adipocyte Adipoq-Cre | Cold, HFD | Increased prostaglandin (PG) production by COX-2 and promotes differentiation of progenitor cells to beige adipocytes | [80] |
| 6 | Raptor | Global S791A knock-in | GLP-1R agonist | Partially resistant to GLP-1R agonist liraglutide-induced weight loss, lesser reduction in fat mass, lower energy expenditure | [62] |
| 7 | Rictor | Adipocyte Fabp4/aP2-Cre | HFD | Increased body (non-adipose organ) size, hyperinsulinemia but glucose-tolerant, elevated levels of IGF1 | [81] |
| 8 | Rictor | Adipocyte Adipoq-Cre | HFD | Normal body growth, insulin intolerance, impaired adipose insulin signaling results in reduced glucose uptake and de novo lipogenesis | [82] |
| 9 | Rictor | Brown adipocyte Myf5-Cre | HFD | Decreased fat mass and adipocyte size, decreased lipogenesis but elevated mitochondrial activity in brown fat, protected against obesity and metabolic disease | [83] |
| 10 | Rictor | Brown adipocyte Ucp1-Cre | Cold, βAR agonist, HFD | Increased cold tolerance, inhibits de novo lipid synthesis, promotes lipid catabolism and thermogenesis, and protects against diet-induced obesity | [84] |
| Liver | |||||
| 11 | Raptor | Hepatocyte Albumin-Cre | Western diet | Gained less weight, reduced fasted liver size, suppresses hepatic de novo lipogenesis, protects mice from hepatic steatosis and hypercholesterolemia | [98] |
| 12 | Raptor | Hepatocyte AAV-TBG-Cre | Fasting and refeeding | Smaller liver with increased steatosis, higher hepatic TAG and decreased TAG secretion under fasting conditions | [113] |
| 13 | Raptor | Hepatocyte Albumin-Cre | HFD, hepatic carcinogen | Increased liver injuries, aberrant regeneration, enhanced fibrosis, inflammation, and hepatocarcinogenesis | [114] |
| 14 | Rictor | Hepatocyte Albumin-Cre | HFD | Smaller liver with lower glycogen and TG content, hepatic insulin resistance, dysregulated hepatic gluconeogenesis, glycolysis and de novo lipogenesis | [118] |
| 15 | Rictor | Hepatocyte Albumin-Cre | HFD | Failed to inhibit hepatic glucose output, glucose intolerance and insulin resistance, resistant to hepatic steatosis on a high-fat diet | [119] |
| Heart | |||||
| 16 | Raptor | Cardiomyocyte MHC-Cre | TAC | Acute cardiac dysfunction in response to overload, metabolic switch from fatty acid to glucose oxidation, abnormal mitochondria, increased apoptosis, and autophagy | [126] |
| 17 | Raptor | Cardiomyocyte MHC-Cre heterozygote | TAC | Attenuates heart failure induced by pressure overload or Gαq overexpression | [130] |
| 18 | Rictor | Cardiomyocyte MHC-Cre | TAC | Accelerates cardiac dysfunction after aortic constriction, reduces PKC protein levels, does not affect hypertrophy, fibrosis, or metabolic gene expression | [138] |
| 19 | Rictor | Cardiomyocyte AAV9 shRNA | MI | Increases myocardial damage and remodeling | [140] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mTOR | mechanistic target of rapamycin; |
| RAPTOR | regulatory-associated protein of mTOR; |
| RICTOR | rapamycin-insensitive companion of mTOR; |
| DEPTOR | DEP domain-containing mTOR-interacting protein; |
| mLST8 | mammalian lethal with SEC13 protein 8; |
| PRAS40 | proline-rich AKT substrate of 40 kDa; |
| mSIN1 | mammalian stress-activated protein kinase-interaction protein 1; |
| PROTOR | protein observed with RICTOR; |
| TSC1/2 | tuberous sclerosis complex 1/2; |
| RHEB | Ras homolog mTORC1 binding; |
| RAGs | Ras-related GTPases; |
| IRS | insulin receptor substrate; |
| PI3K | phosphoinositide 3-kinase; |
| PIP3 | phosphatidylinositol 3,4,5-triphosphate; |
| PTEN | phosphatase and tensin homolog; |
| PDK1 | phosphoinositide-dependent protein kinase 1; |
| GRB10 | growth factor receptor bound protein 10; |
| AKT; | protein kinase B; |
| ERK | extracellular signal-regulated kinase; |
| RSK | ribosomal S6 kinase 1; |
| TNF | tumor necrosis factor; |
| IKKβ | inhibitor of nuclear factor kappa-B kinase subunit beta; |
| AMPK | AMP-activated protein kinase; |
| WNT | wingless and lnt-1; |
| GSKβ | glycogen synthase kinase 3β; |
| REDD1 | regulated in development and DNA damage responses 1; |
| p38 MAPK | p38 mitogen-activated protein kinases; |
| MK2 | MAPK activated protein kinase 2; |
| ROS | reactive oxygen species; |
| NO | nitric oxide; |
| ANP | atrial natriuretic peptide; |
| BNP | B-type natriuretic peptide; |
| GPCR | G protein-coupled receptor; |
| GC | guanylyl cyclase; |
| sGC | soluble guanylyl cyclase; |
| cAMP | cyclic adenosine monophosphate; |
| cGMP | cyclic guanosine monophosphate; |
| PKA | cAMP-dependent protein kinase; |
| PKG | cGMP-dependent protein kinase; |
| SIK3 | salt-inducible kinase 3; |
| FKBP12 | FK506-binding protein of 12 kDa; |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PKA | protein kinase A; |
| PKG | protein kinase G; |
| GPCRs | G protein-coupled receptors; |
| EPAC | exchange protein directly activated by cAMP (EPAC); |
| LKB1 | liver kinase B1; |
| PDE | phosphodiesterase; |
| NPs | natriuretic peptides; |
| TBG | thyroxine binding globulin; |
| TAC | transverse aortic constriction; |
| MHC | α-myosin heavy chain; |
| MI | myocardial infarction; |
| IBMX | 3-Isobutyl-1-methylxanthine; |
| MEFs | Mouse embryonic fibroblast; |
| PTH/PTHrP | parathyroid hor-mone/PTH-related peptide; |
| GLP-1 | Glucagon-like peptide-1; |
| GLP-1RA | GLP-1 receptor agonist; |
| KATP channels | ATP-sensitive K+ channels; |
| TSH | thyroid-stimulating hormone; |
| hCG | human chorionic gonadotropin; |
| β-AR | β-adrenergic receptor; |
| NRVMs | neonatal rat ventricular myocytes; |
| ER | endoplasmic reticulum; |
| GPCRs | G protein-coupled receptors. |
| DNL | de novo lipogenesis; |
| NAFLD | nonalcoholic fatty liver disease; |
| NAFL | nonalcoholic fatty liver; |
| NASH | nonalcoholic hepatic steatosis; |
| HCC | hepatocellular carcinoma; |
| UCP1 | uncoupling protein 1; |
| WAT | white adipose tissue. |
References
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.J.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.R.; Schreiber, S.L. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. CB 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. CB 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef]
- Yuan, H.-X.; Guan, K.-L. The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov. 2015, 5, 1127–1129. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Koszegi, Z.; Lanoiselée, Y.; Miljus, T.; O’Brien, S. G protein-coupled receptor-G protein interactions: A single-molecule perspective. Physiol. Rev. 2021, 101, 857–906. [Google Scholar] [CrossRef] [PubMed]
- Monfar, M.; Lemon, K.P.; Grammer, T.C.; Cheatham, L.; Chung, J.; Vlahos, C.J.; Blenis, J. Activation of pp70/85 S6 kinases in interleukin-2-responsive lymphoid cells is mediated by phosphatidylinositol 3-kinase and inhibited by cyclic AMP. Mol. Cell. Biol. 1995, 15, 326–337. [Google Scholar] [CrossRef]
- Scott, P.H.; Lawrence, J.C., Jr. Attenuation of mammalian target of rapamycin activity by increased cAMP in 3T3-L1 adipocytes. J. Biol. Chem. 1998, 273, 34496–34501. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ponuwei, G.A.; Moore, C.E.; Willars, G.B.; Tee, A.R.; Herbert, T.P. cAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell. Signal. 2011, 23, 1927–1935. [Google Scholar] [CrossRef]
- Rocha, A.; Paternot, S.; Coulonval, K.; Dumont, J.E.; Soares, P.; Roger, P.P. Cyclic AMP Inhibits the Proliferation of Thyroid Carcinoma Cell Lines through Regulation of CDK4 Phosphorylation. Mol. Biol. Cell 2008, 19, 4814–4825. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jee, K.; Kim, D.; Koh, H.; Chung, J. Cyclic AMP inhibits Akt activity by blocking the membrane localization of PDK1. J. Biol. Chem. 2001, 276, 12864–12870. [Google Scholar] [CrossRef]
- Mothe-Satney, I.; Gautier, N.; Hinault, C.; Lawrence, J.C.; Van Obberghen, E. In rat hepatocytes glucagon increases mammalian target of rapamycin phosphorylation on serine 2448 but antagonizes the phosphorylation of its downstream targets induced by insulin and amino acids. J. Biol. Chem. 2004, 279, 42628–42637. [Google Scholar] [CrossRef]
- Kimball, S.R.; Siegfried, B.A.; Jefferson, L.S. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J. Biol. Chem. 2004, 279, 54103–54109. [Google Scholar] [CrossRef]
- Baum, J.; Kimball, S.R.; Jefferson, L.S. Glucagon acts in a dominant manner to repress insulin-induced mammalian target of rapamycin complex 1 signaling in perfused rat liver. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E410–E415. [Google Scholar] [CrossRef]
- Csukasi, F.; Duran, I.; Barad, M.; Barta, T.; Gudernova, I.; Trantirek, L.; Martin, J.H.; Kuo, C.Y.; Woods, J.; Lee, H.; et al. The PTH/PTHrP-SIK3 pathway affects skeletogenesis through altered mTOR signaling. Sci. Transl. Med. 2018, 10, eaat9356. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Bultot, L.; Goransson, O. The Salt-Inducible Kinases: Emerging Metabolic Regulators. Trends Endocrinol. Metab. TEM 2018, 29, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Foretz, M.; Fisher, D.E.; Xavier, R.J.; Kronenberg, H.M. Salt-Inducible Kinases: Physiology, Regulation by cAMP, and Therapeutic Potential. Trends Endocrinol. Metab. TEM 2018, 29, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; de Fatima Silva, F.; Liu, D.; Patel, H.U.; Xu, J.; Zhang, W.; Türk, C.; Krüger, M.; Collins, S. Salt-inducible kinase inhibition promotes the adipocyte thermogenic program and adipose tissue browning. Mol. Metab. 2023, 74, 101753. [Google Scholar] [CrossRef]
- Kwon, G.; Marshall, C.A.; Pappan, K.L.; Remedi, M.S.; McDaniel, M.L. Signaling Elements Involved in the Metabolic Regulation of mTOR by Nutrients, Incretins, and Growth Factors in Islets. Diabetes 2004, 53 (Suppl. S3), S225–S232. [Google Scholar] [CrossRef]
- Brennesvik, E.O.; Ktori, C.; Ruzzin, J.; Jebens, E.; Shepherd, P.R.; Jensen, J. Adrenaline potentiates insulin-stimulated PKB activation via cAMP and Epac: Implications for cross talk between insulin and adrenaline. Cell. Signal. 2005, 17, 1551–1559. [Google Scholar] [CrossRef]
- Moore, C.E.; Xie, J.; Gomez, E.; Herbert, T.P. Identification of cAMP-Dependent Kinase as a Third in Vivo Ribosomal Protein S6 Kinase in Pancreatic β-Cells. J. Mol. Biol. 2009, 389, 480–494. [Google Scholar] [CrossRef]
- Blancquaert, S.; Wang, L.; Paternot, S.; Coulonval, K.; Dumont, J.E.; Harris, T.E.; Roger, P.P. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol. Endocrinol. 2010, 24, 1453–1468. [Google Scholar] [CrossRef][Green Version]
- Palaniappan, M.; Menon, K.M. Human chorionic gonadotropin stimulates theca-interstitial cell proliferation and cell cycle regulatory proteins by a cAMP-dependent activation of AKT/mTORC1 signaling pathway. Mol. Endocrinol. 2010, 24, 1782–1793. [Google Scholar] [CrossRef]
- Kim, H.W.; Ha, S.H.; Lee, M.N.; Huston, E.; Kim, H.; Jang, S.K.; Suh, P.-G.; Houslay, M.D.; Ryu, S.H. Cyclic AMP Controls mTOR through Regulation of the Dynamic Interaction between Rheb and Phosphodiesterase 4D. Mol. Cell. Biol. 2010, 30, 5406–5420. [Google Scholar] [CrossRef]
- Liu, D.; Bordicchia, M.; Zhang, C.; Fang, H.; Wei, W.; Li, J.L.; Guilherme, A.; Guntur, K.; Czech, M.P.; Collins, S. Activation of mTORC1 is essential for beta-adrenergic stimulation of adipose browning. J. Clin. Investig. 2016, 126, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ceddia, R.P.; Collins, S. Cardiac natriuretic peptides promote adipose ‘browning’ through mTOR complex-1. Mol. Metab. 2018, 9, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Bordicchia, M.; Liu, D.; Amri, E.Z.; Ailhaud, G.; Dessi-Fulgheri, P.; Zhang, C.; Takahashi, N.; Sarzani, R.; Collins, S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Investig. 2012, 122, 1022–1036. [Google Scholar] [CrossRef]
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Oeing, C.U.; Nakamura, T.; Pan, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Kokkonen-Simon, K.M.; Lin, B.L.; Chen, A.; Zhu, G.; Bedja, D.; et al. PKG1α Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 2020, 127, 522–533. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Li, Y.; Inoki, K.; Vacratsis, P.; Guan, K.L. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J. Biol. Chem. 2003, 278, 13663–13671. [Google Scholar] [CrossRef]
- Cully, M.; Genevet, A.; Warne, P.H.; Treins, C.; Liu, T.; Bastien, J.; Baum, B.; Tapon, N.; Leevers, S.J.; Downward, J. A Role for p38 Stress-Activated Protein Kinase in Regulation of Cell Growth via TORC1. Mol. Cell. Biol. 2009, 30, 481–495. [Google Scholar] [CrossRef]
- Hernández, G.; Lal, H.; Fidalgo, M.; Guerrero, A.; Zalvide, J.; Force, T.; Pombo, C.M. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 2011, 317, 2938–2949. [Google Scholar] [CrossRef] [PubMed]
- González-Terán, B.; López, J.A.; Rodríguez, E.; Leiva, L.; Martínez-Martínez, S.; Bernal, J.A.; Jiménez-Borreguero, L.J.; Redondo, J.M.; Vazquez, J.; Sabio, G. p38γ and δ promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016, 7, 10477. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 Mitogen-Activated Protein Kinase Is the Central Regulator of Cyclic AMP-Dependent Transcription of the Brown Fat Uncoupling Protein 1 Gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Naguro, I.; Okabe, K.; Funatsu, T.; Furutani, S.; Takeda, K.; Ichijo, H. ASK1 signalling regulates brown and beige adipocyte function. Nat. Commun. 2016, 7, 11158. [Google Scholar] [CrossRef] [PubMed]
- Komalavilas, P.; Shah, P.K.; Jo, H.; Lincoln, T.M. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 34301–34309. [Google Scholar] [CrossRef]
- Doronzo, G.; Viretto, M.; Russo, I.; Mattiello, L.; Di Martino, L.; Cavalot, F.; Anfossi, G.; Trovati, M. Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells: Influence of insulin resistance and oxidative stress. Atherosclerosis 2011, 216, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.N.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Chen, C.H.; Shaikenov, T.; Peterson, T.R.; Aimbetov, R.; Bissenbaev, A.K.; Lee, S.W.; Wu, J.; Lin, H.K.; Sarbassov, D.D. ER Stress Inhibits mTORC2 and Akt Signaling through GSK-3β–Mediated Phosphorylation of Rictor. Sci. Signal. 2011, 4, ra10. [Google Scholar] [CrossRef]
- Koo, J.; Wu, X.; Mao, Z.; Khuri, F.R.; Sun, S.-Y. Rictor Undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal Degradation. J. Biol. Chem. 2015, 290, 14120–14129. [Google Scholar] [CrossRef]
- Urbanska, M.; Gozdz, A.; Macias, M.; Cymerman, I.A.; Liszewska, E.; Kondratiuk, I.; Devijver, H.; Lechat, B.; Van Leuven, F.; Jaworski, J. GSK3β Controls mTOR and Prosurvival Signaling in Neurons. Mol. Neurobiol. 2017, 55, 6050–6062. [Google Scholar] [CrossRef]
- Le, T.D.V.; Liu, D.; Ellis, B.J.; Collins, S.; Ayala, J.E. Glucagon-Like Peptide-1 Receptor Activation Stimulates PKA-Mediated Phosphorylation of Raptor and this Contributes to the Weight Loss Effects of Liraglutide. bioRxiv 2022. [Google Scholar] [CrossRef]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R. American Association of Clinical Endocrinologists and American College of Endocrinology Comprehensive Clinical Practice Guidelines for Medical Care of Patients with Obesity. Endocr. Pract. 2016, 22 (Suppl. S3), 1–203. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S. Obesity wars: Molecular progress confronts an expanding epidemic. Cell 2004, 116, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef]
- Shah, O.J.; Wang, Z.-Y.; Hunter, T. Inappropriate Activation of the TSC/Rheb/mTOR/S6K Cassette Induces IRS1/2 Depletion, Insulin Resistance, and Cell Survival Deficiencies. Curr. Biol. CB 2004, 14, 1650–1656. [Google Scholar] [CrossRef]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of mTORC1-Mediated Inhibition of Growth Factor Signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef]
- Yu, Y.; Yoon, S.-O.; Poulogiannis, G.; Yang, Q.; Max, X.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Wick, K.R.; Werner, E.D.; Langlais, P.; Ramos, F.J.; Dong, L.Q.; Shoelson, S.E.; Liu, F. Grb10 inhibits insulin-stimulated insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase/Akt signaling pathway by disrupting the association of IRS-1/IRS-2 with the insulin receptor. J. Biol. Chem. 2003, 278, 8460–8467. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bai, J.; He, S.; Villarreal, R.; Hu, D.; Zhang, C.; Yang, X.; Liang, H.; Slaga, T.J.; Yu, Y.; et al. Grb10 promotes lipolysis and thermogenesis by phosphorylation-dependent feedback inhibition of mTORC1. Cell Metab. 2014, 19, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Russell, S.J.; Ussar, S.; Boucher, J.; Vernochet, C.; Mori, M.A.; Smyth, G.; Rourk, M.; Cederquist, C.; Rosen, E.D.; et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 2013, 62, 864–874. [Google Scholar] [CrossRef]
- Mullican, S.E.; Tomaru, T.; Gaddis, C.A.; Peed, L.C.; Sundaram, A.; Lazar, M.A. A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol. Endocrinol. 2013, 27, 127–134. [Google Scholar] [CrossRef]
- Jeffery, E.; Berry, R.; Church, C.D.; Yu, S.; Shook, B.A.; Horsley, V.; Rosen, E.D.; Rodeheffer, M.S. Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte 2014, 3, 206–211. [Google Scholar] [CrossRef]
- Lee, P.L.; Tang, Y.; Li, H.; Guertin, D.A. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 2016, 5, 422–432. [Google Scholar] [CrossRef]
- Paolella, L.M.; Mukherjee, S.; Tran, C.M.; Bellaver, B.; Hugo, M.; Luongo, T.S.; Shewale, S.V.; Lu, W.; Chellappa, K.; Baur, J.A. mTORC1 restrains adipocyte lipolysis to prevent systemic hyperlipidemia. Mol. Metab. 2020, 32, 136–147. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, Y.; Wang, C.; Ding, X.; Yang, X.; Wu, D.; Silva, F.; Yang, Z.; Zhou, Q.; Wang, L.; et al. Adipose mTORC1 Suppresses Prostaglandin Signaling and Beige Adipogenesis via the CRTC2-COX-2 Pathway. Cell Rep. 2018, 24, 3180–3193. [Google Scholar] [CrossRef]
- Cybulski, N.; Polak, P.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc. Natl. Acad. Sci. USA 2009, 106, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wallace, M.; Sanchez-Gurmaches, J.; Hsiao, W.Y.; Li, H.; Lee, P.L.; Vernia, S.; Metallo, C.M.; Guertin, D.A. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun. 2016, 7, 11365. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.M.; Calejman, C.M.; Sanchez-Gurmaches, J.; Li, H.; Clish, C.B.; Hettmer, S.; Wagers, A.J.; Guertin, D.A. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. 2014, 8, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Hung, C.M.; Hildebrand, S.R.; Sanchez-Gurmaches, J.; Martinez-Pastor, B.; Gengatharan, J.M.; Wallace, M.; Mukhopadhyay, D.; Martinez Calejman, C.; Luciano, A.K.; et al. Non-canonical mTORC2 Signaling Regulates Brown Adipocyte Lipid Catabolism through SIRT6-FoxO1. Mol. Cell 2019, 75, 807–822.e8. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Müller, T.D.; Blüher, M.; Tschöp, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef]
- Burmeister, M.A.; Brown, J.D.; Ayala, J.E.; Stoffers, D.A.; Sandoval, D.A.; Seeley, R.J.; Ayala, J.E. The glucagon-like peptide-1 receptor in the ventromedial hypothalamus reduces short-term food intake in male mice by regulating nutrient sensor activity. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E651–E662. [Google Scholar] [CrossRef]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2021, 50, 101143. [Google Scholar] [CrossRef]
- Reeder, S.B.; Sirlin, C.B. Quantification of Liver Fat with Magnetic Resonance Imaging. Magn. Reson. Imaging Clin. N. Am. 2010, 18, 337–357. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.E.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2019, 17, 40–52. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metab. Clin. Exp. 2020, 111, 154170. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef]
- Han, J.; Wang, Y. mTORC1 signaling in hepatic lipid metabolism. Protein Cell 2018, 9, 145–151. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Han, J.; Li, E.; Chen, L.; Zhang, Y.-Y.; Wei, F.; Liu, J.; Deng, H.; Wang, Y. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.T.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.L.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong-A, L.; Patange, S.; Raben, N.; Puertollano, R. The Nutrient-Responsive Transcription Factor TFE3 Promotes Autophagy, Lysosomal Biogenesis, and Clearance of Cellular Debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Yeh, M.M.; Yeung, R.S. Tuberous Sclerosis Complex-1 Deficiency Attenuates Diet-Induced Hepatic Lipid Accumulation. PLoS ONE 2011, 6, e18075. [Google Scholar] [CrossRef]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.; Görgün, C.Z.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.-H.; et al. Akt Stimulates Hepatic SREBP1c and Lipogenesis through Parallel mTORC1-Dependent and Independent Pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Quinn, W.J.; Wan, M.; Shewale, S.V.; Gelfer, R.; Rader, D.J.; Birnbaum, M.J.; Titchenell, P.M. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J. Clin. Investig. 2017, 127, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; Park, E.J.; Taniguchi, K.; Lee, J.H.; Shalapour, S.; Valasek, M.A.; Aghajan, M.; Nakagawa, H.; Seki, E.; Hall, M.N.; et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014, 20, 133–144. [Google Scholar] [CrossRef]
- Gosis, B.S.; Wada, S.; Thorsheim, C.; Li, K.; Jung, S.; Rhoades, J.H.; Yang, Y.; Brandimarto, J.; Li, L.; Uehara, K.; et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 2022, 376, eabf8271. [Google Scholar] [CrossRef] [PubMed]
- Uehara, K.; Sostre-Colón, J.; Gavin, M.; Santoleri, D.; Leonard, K.A.; Jacobs, R.L.; Titchenell, P.M. Activation of Liver mTORC1 Protects against NASH via Dual Regulation of VLDL-TAG Secretion and De Novo Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1625–1647. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Qiang, L.; Hayden, M.S.; Sparling, D.P.; Purcell, N.H.; Pajvani, U.B. mTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun. 2016, 7, 10255. [Google Scholar] [CrossRef]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef]
- Yuan, M.; Pino, E.C.; Wu, L.; Kacergis, M.C.; Soukas, A.A. Identification of Akt-independent Regulation of Hepatic Lipogenesis by Mammalian Target of Rapamycin (mTOR) Complex 2. J. Biol. Chem. 2012, 287, 29579–29588. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. The complex network of mTOR signalling in the heart. Cardiovasc. Res. 2022, 118, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pires, K.M.P.; Whitehead, K.J.; Olsen, C.D.; Wayment, B.; Zhang, Y.C.; Bugger, H.; Ilkun, O.; Litwin, S.E.; Thomas, G.; et al. Mechanistic target of rapamycin (Mtor) is essential for murine embryonic heart development and growth. PLoS ONE 2013, 8, e54221. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Izumo, S. Rapamycin Selectively Inhibits Angiotensin II–Induced Increase in Protein Synthesis in Cardiac Myocytes In Vitro: Potential Role of 70-kD S6 Kinase in Angiotensin II– Induced Cardiac Hypertrophy. Circ. Res. 1995, 77, 1040–1052. [Google Scholar] [CrossRef]
- Zhang, D.; Contu, R.; Latronico, M.V.G.; Zhang, J.L.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef]
- Shende, P.S.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Rüegg, M.A.; et al. Cardiac Raptor Ablation Impairs Adaptive Hypertrophy, Alters Metabolic Gene Expression, and Causes Heart Failure in Mice. Circulation 2011, 123, 1073–1082. [Google Scholar] [CrossRef]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.L.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin Attenuates Load-Induced Cardiac Hypertrophy in Mice. Circulation 2003, 107, 1664–1670. [Google Scholar] [CrossRef]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR Signaling with Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef]
- Buss, S.J.; Muenz, S.; Riffel, J.; Malekar, P.; Hagenmueller, M.; Weiss, C.S.; Bea, F.; Bekeredjian, R.; Schinke-Braun, M.; Izumo, S.; et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 2435–2446. [Google Scholar] [CrossRef]
- Dai, D.-F.; Liu, Y.; Basisty, N.; Karunadharma, P.P.; Dastidar, S.G.; Chiao, Y.A.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; et al. Differential effects of various genetic mouse models of the mechanistic target of rapamycin complex I inhibition on heart failure. GeroScience 2019, 41, 847–860. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Flynn, J.M.; O’Leary, M.N.; Zambataro, C.A.; Academia, E.C.; Presley, M.P.; Garrett, B.J.; Zykovich, A.; Mooney, S.D.; Strong, R.; Rosen, C.J.; et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 2013, 12, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.M.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a Critical Regulator of Autophagy during Myocardial Ischemia: Pathophysiological Implications in Obesity and Metabolic Syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential Roles of GSK-3β During Myocardial Ischemia and Ischemia/Reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Durrant, D.; Ockaili, R.A.; Kukreja, R.C. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J. Mol. Cell. Cardiol. 2012, 53, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion: Roles of AMP-Activated Protein Kinase and Beclin 1 in Mediating Autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Di, R.-M.; Wu, X.; Chang, Z.; Zhao, X.; Feng, Q.; Lu, S.; Luan, Q.; Hemmings, B.A.; Li, X.; Yang, Z. S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phosphorylation of Akt. Biochem. J. 2011, 441, 199–207. [Google Scholar] [CrossRef]
- Shende, P.S.; Xu, L.; Morandi, C.; Pentassuglia, L.; Heim, P.; Lebboukh, S.; Berthonneche, C.; Pedrazzini, T.; Kaufmann, B.A.; Hall, M.N.; et al. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc. Res. 2015, 109, 103–114. [Google Scholar] [CrossRef]
- Lamming, D.W.; Mihaylova, M.M.; Katajisto, P.; Baar, E.L.; Yilmaz, Ö.H.; Hutchins, A.W.; Gultekin, Y.; Gaither, R.P.; Sabatini, D.M. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell 2014, 13, 911–917. [Google Scholar] [CrossRef]
- Völkers, M.; Konstandin, M.H.; Doroudgar, S.; Toko, H.; Quijada, P.; Din, S.; Joyo, A.Y.; Ornelas, L.; Samse, K.; Thuerauf, D.J.; et al. Mechanistic Target of Rapamycin Complex 2 Protects the Heart from Ischemic Damage. Circulation 2013, 128, 2132–2144. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, F.; Collins, S. Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease. Int. J. Mol. Sci. 2023, 24, 11497. https://doi.org/10.3390/ijms241411497
Shi F, Collins S. Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease. International Journal of Molecular Sciences. 2023; 24(14):11497. https://doi.org/10.3390/ijms241411497
Chicago/Turabian StyleShi, Fubiao, and Sheila Collins. 2023. "Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease" International Journal of Molecular Sciences 24, no. 14: 11497. https://doi.org/10.3390/ijms241411497
APA StyleShi, F., & Collins, S. (2023). Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease. International Journal of Molecular Sciences, 24(14), 11497. https://doi.org/10.3390/ijms241411497