

Potential Role of Protein Kinase FAM20C on the Brain in Raine Syndrome, an In Silico Analysis
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Identification of Neurologic Alterations in RNS
2.1.1. Identification of Structural and Functional Brain Defects in Reported RNS Cases
2.1.2. Identification of Reported Pathogenic Variants in RNS Individuals with Neurologic Alterations
2.2. FAM20C Targets and Interactors
2.3. Brain Expression of FAM20C Targets and Interactors
2.4. Brain-Cell-Type Expression Levels of Enriched Brain Genes
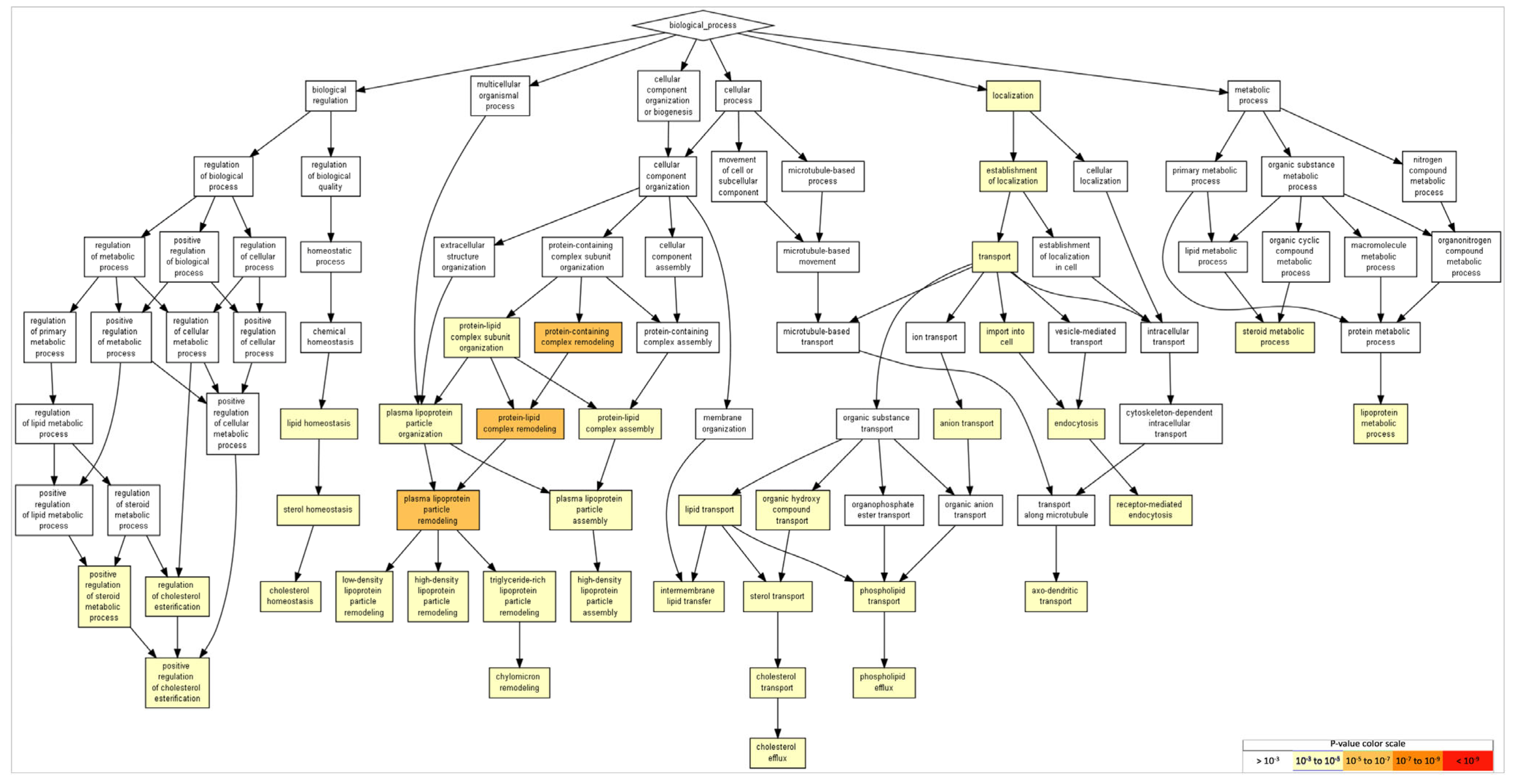
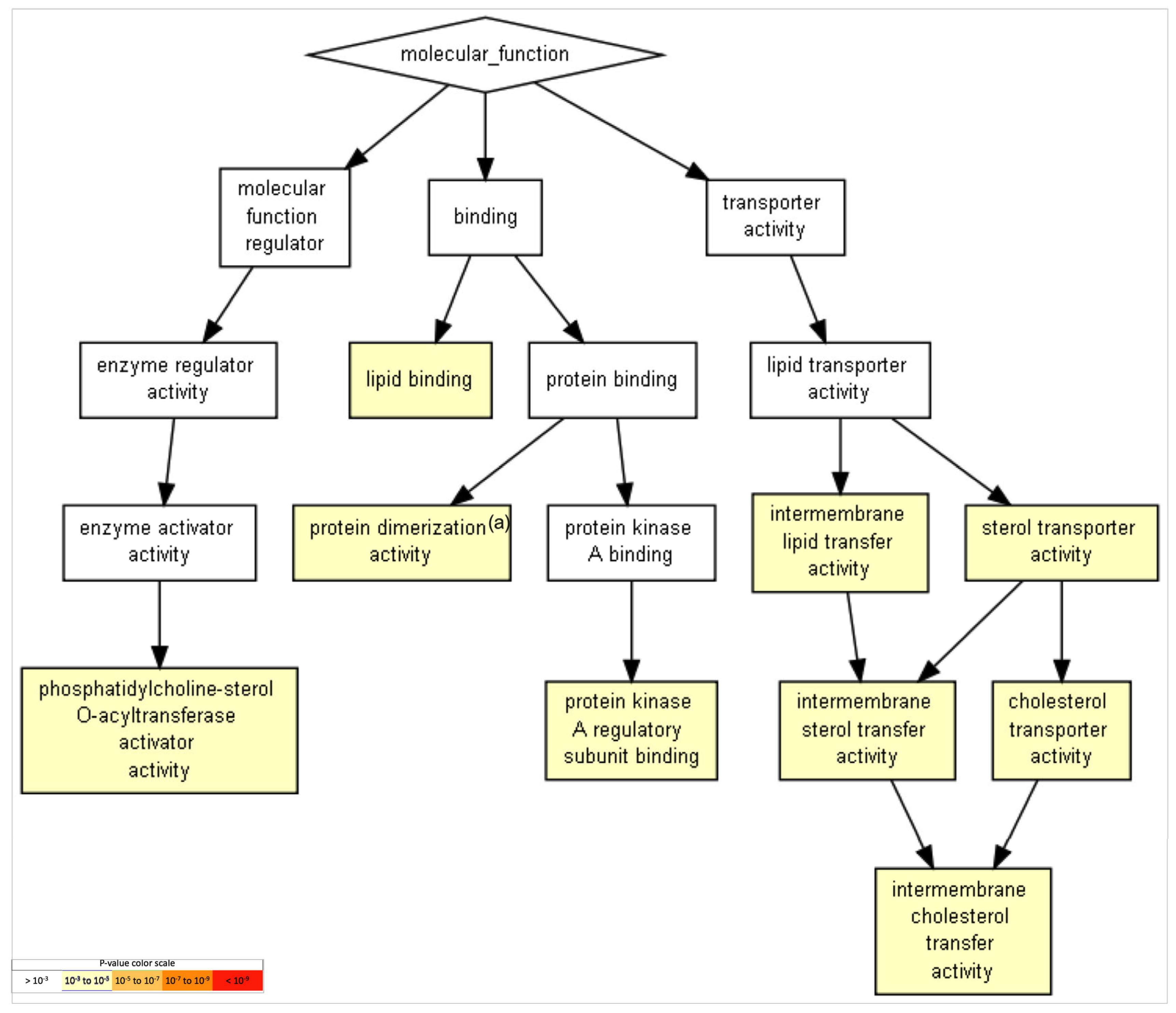
2.5. Gene Ontology of FAM20C Targets–Interactors
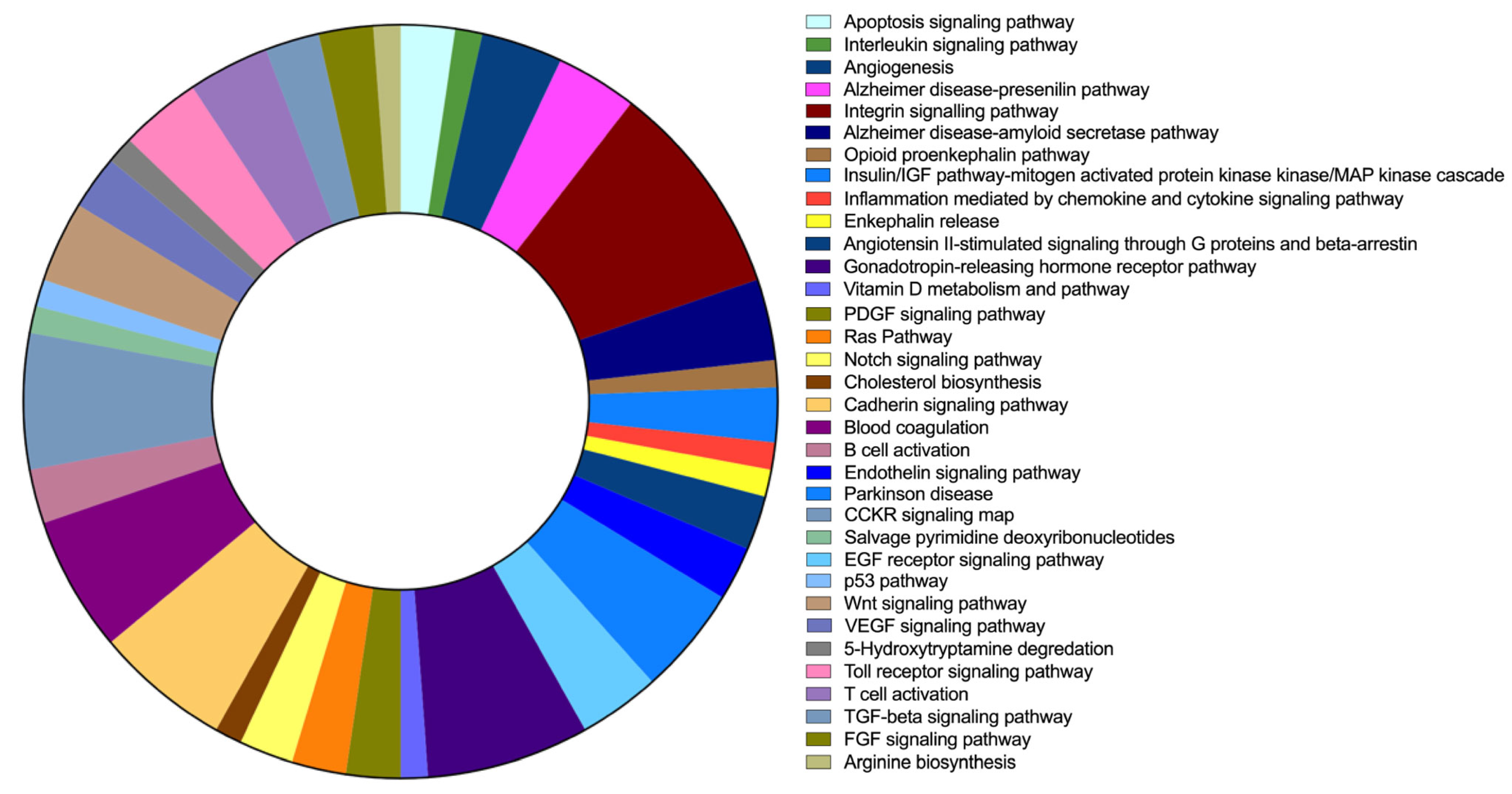
2.6. FAM20C Target–Interactor Pathways
2.7. Brain-Enriched FAM20C Targets–Interactors Associated with Disease
3. Discussion
3.1. Structural and Functional Neurological Defects in RNS
3.1.1. Structural Brain Defects
3.1.2. Functional Brain Defects
3.2. Brain Expression of FAM20C Targets–Interactors
4. Materials and Methods
4.1. Description of Neurologic Alterations in RNS Patients
4.2. Identification of FAM2OC Targets–Interactors
4.3. FAM20 Members and Target–Interactor Expression Levels in the Brain
4.4. Hierarchical Clustering
4.5. Brain-Cell-Type Expression Levels of Enriched Brain Genes
4.6. Gene-Ontology Analysis
4.7. Gene-Pathway Analysis
4.8. Neurological Diseases Associated with FAM20C Targets–Interactors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faundes, V.; Castillo-Taucher, S.; Gonzalez-Hormazabal, P.; Chandler, K.; Crosby, A.; Chioza, B. Raine syndrome: An overview. Eur. J. Med. Genet. 2014, 57, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Sheth, J.; Bhavsar, R.; Gandhi, A.; Sheth, F.; Pancholi, D. A case of Raine syndrome presenting with facial dysmorphy and review of literature. BMC Med. Genet. 2018, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Hsu, R.; Keir, L.S.; Hao, J.; Sivapalan, G.; Ernst, L.M.; Zackai, E.H.; Al-Gazali, L.I.; Hulskamp, G.; Kingston, H.M.; et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am. J. Hum. Genet. 2007, 81, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Brunati, A.M.; Bordin, L.; Clari, G.; James, P.; Quadroni, M.; Baritono, E.; Pinna, L.A.; Donella-Deana, A. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: Identification of primary and secondary phosphorylation sites. Blood 2000, 96, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Palma-Lara, I.; Perez-Ramirez, M.; Garcia Alonso-Themann, P.; Espinosa-Garcia, A.M.; Godinez-Aguilar, R.; Bonilla-Delgado, J.; Lopez-Ornelas, A.; Victoria-Acosta, G.; Olguin-Garcia, M.G.; Moreno, J.; et al. FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes. Int. J. Mol. Sci. 2021, 22, 8039. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wen, J.; Wiley, S.E.; Worby, C.A.; Kinch, L.N.; Xiao, J.; Grishin, N.V.; Dixon, J.E. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 2012, 336, 1150–1153. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Jani, P.; Wang, X.; Lu, Y.; Li, N.; Xiao, J.; Qin, C. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J. Cell. Physiol. 2018, 233, 3476–3486. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Kaewgahya, M.; Khemaleelakul, U.; Dejkhamron, P.; Sutthimethakorn, S.; Thongboonkerd, V.; Iamaroon, A. Enamel-renal-gingival syndrome and FAM20A mutations. Am. J. Med. Genet. Part A 2014, 164, 1–9. [Google Scholar] [CrossRef]
- Rolvien, T.; Kornak, U.; Schinke, T.; Amling, M.; Oheim, R. A novel FAM20C mutation causing hypophosphatemic osteomalacia with osteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of the knee. Osteoporos. Int. 2019, 30, 685–689. [Google Scholar] [CrossRef]
- Koike, T.; Izumikawa, T.; Tamura, J.; Kitagawa, H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 2009, 421, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef]
- Wen, J.; Xiao, J.; Rahdar, M.; Choudhury, B.P.; Cui, J.; Taylor, G.S.; Esko, J.D.; Dixon, J.E. Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 15723–15728. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Murakami, H.; Enomoto, Y.; Tsurusaki, Y.; Takahashi, K.; Mitsuzuka, K.; Ishimoto, H.; Nishimura, G.; Kurosawa, K. A novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling Desbuquois dysplasia. Clin. Genet. 2019, 95, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Staines, K.A.; MacRae, V.E.; Farquharson, C. The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodelling. J. Endocrinol. 2012, 214, 241–255. [Google Scholar] [CrossRef]
- Sreelatha, A.; Kinch, L.N.; Tagliabracci, V.S. The secretory pathway kinases. Biochim. Biophys. Acta 2015, 1854, 1687–1693. [Google Scholar] [CrossRef]
- Das, D.S. Raine Syndrome: A rare case presentation. J. Med. Sci. Clin. Invent. 2017, 4, 3304–3306. [Google Scholar] [CrossRef]
- Kan, A.E.; Kozlowski, K. New distinct lethal osteosclerotic bone dysplasia (Raine syndrome). Am. J. Med Genet. 1992, 43, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Al Mane, K.; Al-Dayel, F.; McDonald, P. Intracranial calcification in Raine syndrome: Radiological pathological correlation. Pediatr. Radiol. 1998, 28, 820–823. [Google Scholar] [CrossRef]
- Chitayat, D.; Shannon, P.; Keating, S.; Toi, A.; Blaser, S.; Friedberg, T.; Superti-Furga, A.; Chong, K.; Unger, S. Raine syndrome: A rare lethal osteosclerotic bone dysplasia. Prenatal diagnosis, autopsy, and neuropathological findings. Am. J. Med Genet. Part A 2007, 143A, 3280–3285. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, F.K.; AlSwaid, A.; Youssef, T.; Al Azzawi, M.; Crosby, A.; AlBalwi, M.A. Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am. J. Med Genet. Part A 2013, 161, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; McAlister, W.H.; Fallon, M.D.; Pierpont, M.E.; Bijanki, V.N.; Duan, S.; A Otaify, G.; Sly, W.S.; Mumm, S. Raine Syndrome (OMIM #259775), Caused By FAM20C Mutation, Is Congenital Sclerosing Osteomalacia With Cerebral Calcification (OMIM 259660). J. Bone Miner. Res. 2016, 32, 757–769. [Google Scholar] [CrossRef]
- Hernández-Zavala, A.; Cortés-Camacho, F.; Palma-Lara, I.; Godínez-Aguilar, R.; Espinosa, A.M.; Pérez-Durán, J.; Villanueva-Ocampo, P.; Ugarte-Briones, C.; Serrano-Bello, C.A.; Sánchez-Santiago, P.J.; et al. Two Novel FAM20C Variants in a Family with Raine Syndrome. Genes 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Seidahmed, M.Z.; Alazami, A.M.; Abdelbasit, O.; Al Hussein, K.; Miqdad, A.M.; Abu-Sa’Da, O.; Mustafa, T.; Bahjat, S.; Alkuraya, F.S. Report of a case of Raine syndrome and literature review. Am. J. Med Genet. Part A 2015, 167, 2394–2398. [Google Scholar] [CrossRef]
- Hung, C.Y.; Rodriguez, M.; Roberts, A.; Bauer, M.; Mihalek, I.; Bodamer, O. A novel FAM20C mutation causes a rare form of neonatal lethal Raine syndrome. Am. J. Med Genet. Part A 2019, 179, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- El-Dessouky, S.H.; Abdel-Hamid, M.S.; Abdel-Ghafar, S.F.; Aboulghar, M.M.; Gaafar, H.M.; Fouad, M.; Ahmed, A.H.; Abdel-Salam, G.M.H. Raine syndrome: Prenatal diagnosis based on recognizable fetal facial features and characteristic intracranial calcification. Prenat. Diagn. 2020, 40, 1578–1597. [Google Scholar] [CrossRef]
- Bajaj, S.; Nabi, F.; Shah, J.; Sheth, H. Recurrent variant c.1680C > A in FAM20C gene and genotype-phenotype correlation in a patient with Raine syndrome: A case report. BMC Pediatr. 2021, 21, 1–5. [Google Scholar] [CrossRef]
- Simpson, M.; Scheuerle, A.; Hurst, J.; A Patton, M.; Stewart, H.; Crosby, A.H. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin. Genet. 2009, 75, 271–276. [Google Scholar] [CrossRef]
- Rafaelsen, S.H.; Raeder, H.; Fagerheim, A.K.; Knappskog, P.; O Carpenter, T.; Johansson, S.; Bjerknes, R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J. Bone Miner. Res. 2013, 28, 1378–1385. [Google Scholar] [CrossRef]
- Acevedo, A.C.; Poulter, J.A.; Alves, P.G.; De Lima, C.L.; Castro, L.C.; Yamaguti, P.M.; Paula, L.M.; Parry, D.A.; Logan, C.V.; Smith, C.E.L.; et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med Genet. 2015, 16, 1–11. [Google Scholar] [CrossRef]
- Elalaoui, S.C.; Al-Sheqaih, N.; Ratbi, I.; Urquhart, J.E.; O’Sullivan, J.; Bhaskar, S.; Williams, S.S.; Elalloussi, M.; Lyahyai, J.; Sbihi, L.; et al. Non lethal Raine syndrome and differential diagnosis. Eur. J. Med Genet. 2016, 59, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Tada, K.; Takeuchi, A.; Nakamura, M.; Marunaka, H.; Washio, Y.; Tanaka, H.; Miya, F.; Okamoto, N.; Kageyama, M. Fetal ultrasonographic findings including cerebral hyperechogenicity in a patient with non-lethal form of Raine syndrome. Am. J. Med Genet. Part A 2018, 176, 682–686. [Google Scholar] [CrossRef]
- Mameli, C.; Zichichi, G.; Mahmood, N.; Elalaoui, S.C.; Mirza, A.; Dharmaraj, P.; Zuccotti, G. Natural history of non-lethal Raine syndrome during childhood. Orphanet J. Rare Dis. 2020, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eltan, M.; Alavanda, C.; Abali, Z.Y.; Ergenekon, P.; Ozturk, N.Y.; Sakar, M.; Dagcinar, A.; Kirkgoz, T.; Kaygusuz, S.B.; Gokdemir, Y.; et al. A Rare Cause of Hypophosphatemia: Raine Syndrome Changing Clinical Features with Age. Calcif. Tissue Int. 2020, 107, 96–103. [Google Scholar] [CrossRef]
- Hirst, L.; Abou-Ameira, G.; Critchlow, S. Hypophosphataemic Rickets Secondary to Raine Syndrome: A Review of the Literature and Case Reports of Three Paediatric Patients’ Dental Management. Case Rep. Pediatr. 2021, 2021, 1–6. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Wang, P.; Li, X.; Zhang, S.; Tian, M.; Dai, Y.; Fu, L. Systematic network-based discovery of a Fam20C inhibitor (FL-1607) with apoptosis modulation in triple-negative breast cancer. Mol. Biosyst. 2016, 12, 2108–2118. [Google Scholar] [CrossRef]
- Baranano, K.; Burd, I. CNS Malformations in the Newborn. Matern. Health Neonatol. Perinatol. 2022, 8, 1. [Google Scholar] [CrossRef]
- Hehr, U.; Schuierer, G. Genetic assessment of cortical malformations. Neuropediatrics 2011, 42, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef] [PubMed]
- Mirzaa, G.M.; Millen, K.J.; Barkovich, A.J.; Dobyns, W.B.; Paciorkowski, A.R. The Developmental Brain Disorders Database (DBDB): A curated neurogenetics knowledge base with clinical and research applications. Am. J. Med. Genet. Part A 2014, 164, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Paciorkowski, A.R.; Weisenberg, J.; Kelley, J.B.; Spencer, A.; Tuttle, E.; Ghoneim, D.; Thio, L.L.; Christian, S.L.; Dobyns, W.B.; Paschal, B.M. Autosomal recessive mutations in nuclear transport factor KPNA7 are associated with infantile spasms and cerebellar malformation. Eur. J. Hum. Genet. 2014, 22, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Duque, A.; Krsnik, Z.; Kostovic, I.; Rakic, P. Secondary expansion of the transient subplate zone in the developing cerebrum of human and nonhuman primates. Proc. Natl. Acad. Sci. USA 2016, 113, 9892–9897. [Google Scholar] [CrossRef]
- Rash, B.G.; Ackman, J.B.; Rakic, P. Bidirectional radial Ca2+ activity regulates neurogenesis and migration during early cortical column formation. Sci. Adv. 2016, 2, e1501733. [Google Scholar] [CrossRef]
- Baala, L.; Briault, S.; Etchevers, H.C.; Laumonnier, F.; Natiq, A.; Amiel, J.; Boddaert, N.; Picard, C.; Sbiti, A.; Asermouh, A.; et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007, 39, 454–456. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef]
- Colantuoni, C.; Lipska, B.K.; Ye, T.; Hyde, T.M.; Tao, R.; Leek, J.T.; Colantuoni, E.A.; Elkahloun, A.G.; Herman, M.M.; Weinberger, D.R.; et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011, 478, 519–523. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef]
- Jaffe, A.E. Postmortem human brain genomics in neuropsychiatric disorders—How far can we go? Curr. Opin. Neurobiol. 2016, 36, 107–111. [Google Scholar] [CrossRef]
- Pletikos, M.; Sousa, A.M.; Sedmak, G.; Meyer, K.A.; Zhu, Y.; Cheng, F.; Li, M.; Kawasawa, Y.I.; Sestan, N. Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron 2014, 81, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Florio, M.; Albert, M.; Taverna, E.; Namba, T.; Brandl, H.; Lewitus, E.; Haffner, C.; Sykes, A.; Wong, F.K.; Peters, J.; et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 2015, 347, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Bhaduri, A.; Pollen, A.A.; Alvarado, B.; Mostajo-Radji, M.A.; Di Lullo, E.; Haeussler, M.; Sandoval-Espinosa, C.; Liu, S.J.; Velmeshev, D.; et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 2017, 358, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef]
- Van Essen, M.J.; Nayler, S.; Becker, E.B.E.; Jacob, J. Deconstructing cerebellar development cell by cell. PLoS Genet. 2020, 16, e1008630. [Google Scholar] [CrossRef]
- Ten Donkelaar, H.J.; Lammens, M.; Wesseling, P.; Thijssen, H.O.; Renier, W.O. Development and developmental disorders of the human cerebellum. J. Neurol. 2003, 250, 1025–1036. [Google Scholar] [CrossRef]
- Rahimi, B.; Makis, W.; Riauka, T.A.; McEwan, A.J.; Morrish, D. 131I-MIBG Therapy in a Metastatic Small Bowel Neuroendocrine Tumor Patient Undergoing Hemodialysis. Clin. Nucl. Med. 2017, 42, 350–353. [Google Scholar] [CrossRef]
- Amore, G.; Spoto, G.; Ieni, A.; Vetri, L.; Quatrosi, G.; Di Rosa, G.; Nicotera, A.G. A Focus on the Cerebellum: From Embryogenesis to an Age-Related Clinical Perspective. Front. Syst. Neurosci. 2021, 15, 646052. [Google Scholar] [CrossRef]
- Schmahmann, J.D. The cerebellum and cognition. Neurosci. Lett. 2019, 688, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J.; Schmahmann, J.D. Functional topography of the human cerebellum. Handb. Clin. Neurol. 2018, 154, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bodranghien, F.; Bastian, A.; Casali, C.; Hallett, M.; Louis, E.D.; Manto, M.; Marien, P.; Nowak, D.A.; Schmahmann, J.D.; Serrao, M.; et al. Consensus Paper: Revisiting the Symptoms and Signs of Cerebellar Syndrome. Cerebellum 2016, 15, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Tam, E.W.Y.; Chau, V.; Lavoie, R.; Chakravarty, M.M.; Guo, T.; Synnes, A.; Zwicker, J.; Grunau, R.; Miller, S.P. Neurologic Examination Findings Associated With Small Cerebellar Volumes After Prematurity. J. Child Neurol. 2019, 34, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.L.; Bae, D.; Kiyatkin, E.A. Relationships between locomotor activation and alterations in brain temperature during selective blockade and stimulation of dopamine transmission. Neuroscience 2007, 145, 335–343. [Google Scholar] [CrossRef]
- Folliot-Le Doussal, L.; Chadie, A.; Brasseur-Daudruy, M.; Verspyck, E.; Saugier-Veber, P.; Marret, S.; Perinatal Network of, H.-N. Neurodevelopmental outcome in prenatally diagnosed isolated agenesis of the corpus callosum. Early Hum. Dev. 2018, 116, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.J.; Sherr, E.H.; Barkovich, A.J.; Richards, L.J. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014, 137, 1579–1613. [Google Scholar] [CrossRef]
- Kidron, D.; Shapira, D.; Ben Sira, L.; Malinger, G.; Lev, D.; Cioca, A.; Sharony, R.; Lerman Sagie, T. Agenesis of the corpus callosum. An autopsy study in fetuses. Virchows Arch. 2016, 468, 219–230. [Google Scholar] [CrossRef]
- Hofman, J.; Hutny, M.; Sztuba, K.; Paprocka, J. Corpus Callosum Agenesis: An Insight into the Etiology and Spectrum of Symptoms. Brain Sci. 2020, 10, 625. [Google Scholar] [CrossRef]
- Chadie, A.; Radi, S.; Trestard, L.; Charollais, A.; Eurin, D.; Verspyck, E.; Marret, S.; Haute-Normandie Perinatal, N. Neurodevelopmental outcome in prenatally diagnosed isolated agenesis of the corpus callosum. Acta Paediatr. 2008, 97, 420–424. [Google Scholar] [CrossRef]
- D’Antonio, F.; Pagani, G.; Familiari, A.; Khalil, A.; Sagies, T.L.; Malinger, G.; Leibovitz, Z.; Garel, C.; Moutard, M.L.; Pilu, G.; et al. Outcomes Associated With Isolated Agenesis of the Corpus Callosum: A Meta-analysis. Pediatrics 2016, 138, e20160445. [Google Scholar] [CrossRef] [PubMed]
- Des Portes, V.; Rolland, A.; Velazquez-Dominguez, J.; Peyric, E.; Cordier, M.P.; Gaucherand, P.; Massardier, J.; Massoud, M.; Curie, A.; Pellot, A.S.; et al. Outcome of isolated agenesis of the corpus callosum: A population-based prospective study. Eur. J. Paediatr. Neurol. 2018, 22, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Bellman, M.; Byrne, O.; Sege, R. Developmental assessment of children. BMJ 2013, 346, e8687. [Google Scholar] [CrossRef] [PubMed]
- Belanger, S.A.; Caron, J. Evaluation of the child with global developmental delay and intellectual disability. Paediatr. Child Health 2018, 23, 403–419. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef]
- Funk, L.; Su, K.C.; Ly, J.; Feldman, D.; Singh, A.; Moodie, B.; Blainey, P.C.; Cheeseman, I.M. The phenotypic landscape of essential human genes. Cell 2022, 185, 4634–4653.e4622. [Google Scholar] [CrossRef]
- Liao, E.N.; Li, Y.; Fung, A.; Lawless, L.; Czechowicz, J.; Ho, M.; Luu, K.; Meyer, L.; Mohamad, N.I.; Nadaraja, G.S.; et al. Predictors of successful natural sleep MRI for sensorineural hearing loss in infants. Int. J. Pediatr. Otorhinolaryngol. 2023, 165, 111430. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Smith, R.J. Navigating genetic diagnostics in patients with hearing loss. Curr. Opin. Pediatr. 2016, 28, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, A.M.; Ishai, R.; McKenna, M.J. Otosclerosis: Temporal Bone Pathology. Otolaryngol. Clin. N. Am. 2018, 51, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, I.; Valgaeren, H.; Tomas-Roca, L.; Sommen, M.; Altunoglu, U.; Wesdorp, M.; Beyens, M.; Fransen, E.; Nasir, A.; Vandeweyer, G.; et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet. Med. 2019, 21, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Chelse, A.B.; Kelley, K.; Hageman, J.R.; Koh, S. Initial evaluation and management of a first seizure in children. Pediatr. Ann. 2013, 42, 244–248. [Google Scholar] [CrossRef]
- Camfield, P.; Camfield, C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic. Disord. 2015, 17, 117–123. [Google Scholar] [CrossRef]
- Volpe, J.J. Postnatal sepsis, necrotizing entercolitis, and the critical role of systemic inflammation in white matter injury in premature infants. J. Pediatr. 2008, 153, 160–163. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18024. [Google Scholar] [CrossRef]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef]
- Lazarov, O.; Demars, M.P. All in the Family: How the APPs Regulate Neurogenesis. Front. Neurosci. 2012, 6, 81. [Google Scholar] [CrossRef]
- Essa, H.; Peyton, L.; Hasan, W.; Leon, B.E.; Choi, D.S. Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Cells 2022, 11, 286. [Google Scholar] [CrossRef]
- Klein, S.; Goldman, A.; Lee, H.; Ghahremani, S.; Bhakta, V.; Center, U.C.G.; Nelson, S.F.; Martinez-Agosto, J.A. Truncating mutations in APP cause a distinct neurological phenotype. Ann. Neurol. 2016, 80, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Malicdan, M.C.V.; Wang, J.A.; Pri-Chen, H.; Hess, R.A.; Fischer, R.; O’Brien, K.J.; Merideth, M.A.; Gahl, W.A.; Gochuico, B.R. Hermansky-Pudlak syndrome: Mutation update. Hum. Mutat. 2020, 41, 543–580. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Z.; Zhong, W.; Yang, H.; Li, Y. A study of 285 cases of cranial vault suture closure in Chinese adults. Surg. Radiol. Anat. 2022, 44, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Shen, X.; Kang, Q.; Kuang, X.; Ning, Z.; Liu, S.; Liao, H.; Cao, Z.; Yang, L. Clinical and Genetic Study on a Chinese Patient with Infantile Onset Epileptic Encephalopathy carrying a PPP3CA Null Variant: A case report. BMC Pediatr. 2020, 20, 315. [Google Scholar] [CrossRef]
- Despa, S.; Lingrel, J.B.; Bers, D.M. Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc. Res. 2012, 95, 480–486. [Google Scholar] [CrossRef]
- Hartford, A.K.; Messer, M.L.; Moseley, A.E.; Lingrel, J.B.; Delamere, N.A. Na,K-ATPase alpha 2 inhibition alters calcium responses in optic nerve astrocytes. Glia 2004, 45, 229–237. [Google Scholar] [CrossRef]
- Lafreniere, R.G.; Rouleau, G.A. Migraine: Role of the TRESK two-pore potassium channel. Int. J. Biochem. Cell Biol. 2011, 43, 1533–1536. [Google Scholar] [CrossRef]
- Chatron, N.; Cabet, S.; Alix, E.; Buenerd, A.; Cox, P.; Guibaud, L.; Labalme, A.; Marks, P.; Osio, D.; Putoux, A.; et al. A novel lethal recognizable polymicrogyric syndrome caused by ATP1A2 homozygous truncating variants. Brain 2019, 142, 3367–3374. [Google Scholar] [CrossRef]
- Key, J.; Mueller, A.K.; Gispert, S.; Matschke, L.; Wittig, I.; Corti, O.; Munch, C.; Decher, N.; Auburger, G. Ubiquitylome profiling of Parkin-null brain reveals dysregulation of calcium homeostasis factors ATP1A2, Hippocalcin and GNA11, reflected by altered firing of noradrenergic neurons. Neurobiol. Dis. 2019, 127, 114–130. [Google Scholar] [CrossRef]
- Chin, J.H.; Tse, F.W.; Harris, K.; Jhamandas, J.H. Beta-amyloid enhances intracellular calcium rises mediated by repeated activation of intracellular calcium stores and nicotinic receptors in acutely dissociated rat basal forebrain neurons. Brain Cell Biol. 2006, 35, 173–186. [Google Scholar] [CrossRef]
- Moseley, A.E.; Lieske, S.P.; Wetzel, R.K.; James, P.F.; He, S.; Shelly, D.A.; Paul, R.J.; Boivin, G.P.; Witte, D.P.; Ramirez, J.M.; et al. The Na,K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J. Biol. Chem. 2003, 278, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Takeichi, M. Cadherins in brain morphogenesis and wiring. Physiol. Rev. 2012, 92, 597–634. [Google Scholar] [CrossRef] [PubMed]
- Accogli, A.; St-Onge, J.; Addour-Boudrahem, N.; Lafond-Lapalme, J.; Laporte, A.D.; Rouleau, G.A.; Riviere, J.B.; Srour, M. Heterozygous Missense Pathogenic Variants Within the Second Spectrin Repeat of SPTBN2 Lead to Infantile-Onset Cerebellar Ataxia. J. Child Neurol. 2020, 35, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.; Stavsky, A.; Kadir, R.; Drabkin, M.; Wormser, O.; Yogev, Y.; Dolgin, V.; Proskorovski-Ohayon, R.; Perez, Y.; Nudelman, H.; et al. CDH2 mutation affecting N-cadherin function causes attention-deficit hyperactivity disorder in humans and mice. Nat. Commun. 2021, 12, 6187. [Google Scholar] [CrossRef]
- Horn, M.E.; Nicoll, R.A. Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 589–594. [Google Scholar] [CrossRef]
- Reines, A.; Bernier, L.P.; McAdam, R.; Belkaid, W.; Shan, W.; Koch, A.W.; Seguela, P.; Colman, D.R.; Dhaunchak, A.S. N-cadherin prodomain processing regulates synaptogenesis. J. Neurosci. 2012, 32, 6323–6334. [Google Scholar] [CrossRef]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lubke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Prox, J.; Bernreuther, C.; Altmeppen, H.; Grendel, J.; Glatzel, M.; D’Hooge, R.; Stroobants, S.; Ahmed, T.; Balschun, D.; Willem, M.; et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 2013, 33, 12915–12928. [Google Scholar] [CrossRef]
- Endres, K.; Deller, T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front. Mol. Neurosci. 2017, 10, 56. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Dai, Z.; Meng, T.; Tu, S.; Yan, Y. IGF-1 reduces BACE-1 expression in PC12 cells via activation of PI3-K/Akt and MAPK/ERK1/2 signaling pathways. Neurochem. Res. 2011, 36, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, Y.; Qiao, P.F.; Zhao, F.L.; Yan, Y. PPAR-alpha agonist regulates amyloid-beta generation via inhibiting BACE-1 activity in human neuroblastoma SH-SY5Y cells transfected with APPswe gene. Mol. Cell. Biochem. 2015, 408, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Halliday, G.M.; Garner, B. Apolipoprotein-E forms dimers in human frontal cortex and hippocampus. BMC Neurosci. 2010, 11, 23. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin. Lipidol. 2010, 51, 555–573. [Google Scholar] [CrossRef]
- Pedrini, S.; Chatterjee, P.; Nakamura, A.; Tegg, M.; Hone, E.; Rainey-Smith, S.R.; Rowe, C.C.; Dore, V.; Villemagne, V.L.; Ames, D.; et al. The Association Between Alzheimer’s Disease-Related Markers and Physical Activity in Cognitively Normal Older Adults. Front. Aging Neurosci. 2022, 14, 771214. [Google Scholar] [CrossRef]
- Pedrini, S.; Doecke, J.D.; Hone, E.; Wang, P.; Thota, R.; Bush, A.I.; Rowe, C.C.; Dore, V.; Villemagne, V.L.; Ames, D.; et al. Plasma high-density lipoprotein cargo is altered in Alzheimer’s disease and is associated with regional brain volume. J. Neurochem. 2022, 163, 53–67. [Google Scholar] [CrossRef]
- De Silva, H.V.; Stuart, W.D.; Duvic, C.R.; Wetterau, J.R.; Ray, M.J.; Ferguson, D.G.; Albers, H.W.; Smith, W.R.; Harmony, J.A. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 1990, 265, 13240–13247. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohno, H.; Hasegawa, A.; Toshima, S.; Amaki, T.; Kurabayashi, M.; Nagai, R.; Suzuki, T.; Amaki, T.; Nagai, R.; et al. Diagnostic implications of circulating oxidized low density lipoprotein levels as a biochemical risk marker of coronary artery disease. Clin. Biochem. 2002, 35, 347–353. [Google Scholar] [CrossRef]
- Herring, S.K.; Moon, H.J.; Rawal, P.; Chhibber, A.; Zhao, L. Brain clusterin protein isoforms and mitochondrial localization. eLife 2019, 8, e48255. [Google Scholar] [CrossRef]
- Athanas, K.M.; Mauney, S.L.; Woo, T.W. Increased extracellular clusterin in the prefrontal cortex in schizophrenia. Schizophr. Res. 2015, 169, 381–385. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Polihronis, M.; Paizis, K.; Carter, G.; Sedal, L.; Murphy, B. Elevation of human cerebrospinal fluid clusterin concentration is associated with acute neuropathology. J. Neurol. Sci 1993, 115, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Gennarelli, M.; Russo, I. Leucine-rich repeat kinase 2-related functions in GLIA: An update of the last years. Biochem. Soc. Trans. 2021, 49, 1375–1384. [Google Scholar] [CrossRef]
- Filippini, A.; Mutti, V.; Faustini, G.; Longhena, F.; Ramazzina, I.; Rizzi, F.; Kaganovich, A.; Roosen, D.A.; Landeck, N.; Duffy, M.; et al. Extracellular clusterin limits the uptake of alpha-synuclein fibrils by murine and human astrocytes. Glia 2021, 69, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Llana, O.; Scott, S.A.; Maslen, S.L.; Anderson, J.M.; Boyle, J.; Chowhdury, R.R.; Tyers, P.; Barker, R.A.; Kelly, C.M.; Rosser, A.E.; et al. Clusterin secreted by astrocytes enhances neuronal differentiation from human neural precursor cells. Cell Death Differ. 2011, 18, 907–913. [Google Scholar] [CrossRef]
- Danik, M.; Chabot, J.G.; Hassan-Gonzalez, D.; Suh, M.; Quirion, R. Localization of sulfated glycoprotein-2/clusterin mRNA in the rat brain by in situ hybridization. J. Comp. Neurol. 1993, 334, 209–227. [Google Scholar] [CrossRef]
- Xue, T.; Wei, L.; Zha, D.J.; Qiao, L.; Lu, L.J.; Chen, F.Q.; Qiu, J.H. Exposure to acoustic stimuli promotes the development and differentiation of neural stem cells from the cochlear nuclei through the clusterin pathway. Int. J. Mol. Med. 2015, 35, 637–644. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Bracher, A.; Hartl, F.U. The chaperone Clusterin in neurodegeneration-friend or foe? Bioessays 2022, 44, e2100287. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Woody, S.K.; Zhou, H.; Ibrahimi, S.; Dong, Y.; Zhao, L. Human ApoE varepsilon2 Promotes Regulatory Mechanisms of Bioenergetic and Synaptic Function in Female Brain: A Focus on V-type H+-ATPase. J. Alzheimer’s Dis. 2016, 53, 1015–1031. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Rose, J.; Tulloch, J.; Henstridge, C.; Smith, C.; Spires-Jones, T.L. Clusterin accumulates in synapses in Alzheimer’s disease and is increased in apolipoprotein E4 carriers. Brain Commun. 2019, 1, fcz003. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Emerek, K.; Sorensen, P.L.; Graff, C.; Loring, Z.; Jackson, K.P.; Polcwiartek, C.; Singh, J.P.; Kisslo, J.; Sogaard, P.; et al. Relationship Between Electrical and Mechanical Dyssynchrony and Outcomes Among Patients Undergoing Cardiac Resynchronization Therapy. Circ. Arrhythm. Electrophysiol. 2021, 14, e010217. [Google Scholar] [CrossRef]
- Hok, A.H.Y.S.; Dijkstra, A.A.; Rabano, A.; Hoozemans, J.J.; Castillo, L.; Seelaar, H.; van Swieten, J.C.; Pijnenburg, Y.A.L.; Teunissen, C.E.; Del Campo, M. Apolipoprotein L1 is increased in frontotemporal lobar degeneration post-mortem brain but not in ante-mortem cerebrospinal fluid. Neurobiol. Dis. 2022, 172, 105813. [Google Scholar] [CrossRef] [PubMed]
- Mimmack, M.L.; Ryan, M.; Baba, H.; Navarro-Ruiz, J.; Iritani, S.; Faull, R.L.; McKenna, P.J.; Jones, P.B.; Arai, H.; Starkey, M.; et al. Gene expression analysis in schizophrenia: Reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc. Natl. Acad. Sci. USA 2002, 99, 4680–4685. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; He, Y.; Phan, K.; Ahmed, R.M.; Rye, K.A.; Piguet, O.; Hodges, J.R.; Halliday, G.M. Altered High Density Lipoprotein Composition in Behavioral Variant Frontotemporal Dementia. Front. Neurosci. 2018, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Xu, S.; Tang, C. Cholesterol and Hedgehog Signaling: Mutual Regulation and Beyond. Front. Cell Dev. Biol. 2022, 10, 774291. [Google Scholar] [CrossRef]
- Sjostedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Thomas, P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Sauch, J.; Sanz, F.; Furlong, L.I. The DisGeNET cytoscape app: Exploring and visualizing disease genomics data. Comput. Struct. Biotechnol. J. 2021, 19, 2960–2967. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Lethal/Non-Lethal | Reference | Variant Description | Variant type | KD Effect | Variant Effect | Language Delay | Developmental delay/Learning difficulty | Seizures | Hypoacusia | Hydrocephaly | Visual impairment | Dystonic movements | Hypertonia | Hypotonia | Cortical defects | Corpus callosal Defects | Cerebellum Defects | Non-visualization of pituitary gland | Hypoplastic appearance of posterior part of the brain | White matter defects/Gliosis/astrogliosis | Encephalocele | Cortex | White matter/Glia | Cerebellum | Corpus callosum | Posterior brain | Pineal Gland | Functional only |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L | [19] | ND | - | - | - | r | r | n | n | |||||||||||||||||||
| 2 | L | [20] | ND | - | - | - | R | ||||||||||||||||||||||
| 3 | L | [21] | ND | - | - | - | r | r | n | n | |||||||||||||||||||
| 4 | L | [22] | 46,XY.ar [hg19] 7p22.3 (36480- 523731)x0 | GDel | KDºKDº | CG AM | X | A | |||||||||||||||||||||
| 5 | L | [23] | c.1094G>A(Gly365Asp)/deletion | MS/GDel | KD*KDº | E6 HM/CG AM | X | A | |||||||||||||||||||||
| 6 | L | [23] | c.1094G>A(Gly365Asp)/deletion | MS/GDel | KD*KDº | E6 HM/CG AM | x | a | |||||||||||||||||||||
| 7 | L | [24] | c.456dupC(Gly153Argfs*56)/c.474delC (Ser159Profs*28) | NS/NS | KDºKDº | E1 AM/E1 AM | r | r | n | n | |||||||||||||||||||
| 8 | L | [25] | c.1225C>T(p.Arg409Cys) | MS | KD*KD* | E6 HM | r | r | r | n | n | n | |||||||||||||||||
| 9 | L | [26] | c.1007T>G(p.Met336Arg) | MS | KD**KD** | E5 HM | r | ||||||||||||||||||||||
| 10 | L | [27] | c.456delC(p.Gly153Alafs*34) | NS | KDºKDº | E1 HM | r | n | |||||||||||||||||||||
| 11 | L | [28] | c.1680C >A(p.Cys560Ter) | MS | KD*KD* | E10 HM | x | x | x | a | |||||||||||||||||||
| TOTAL | KDºKDº=3 KD*KD*=2KD**KD**= 2KD*KDº= 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 3 | 3 | 2 | 4 | 0 | 0 | 2 | 2 | 3 | 2 | 4 | 1 | 4 | |||||||
| 1 | NL | [29] | c.1351G>A(p.Asp451Asn) | MS | KD*KD* | E7 HM | X | x | x | x | x | A | |||||||||||||||||
| 2 | NL | [29] | c.773T>A(p.IIe258Asn)/c.838 G>A(p.Gly280Arg) | MS | KD**KD** | E2 HM/E3 HM | X | x | x | x | A | ||||||||||||||||||
| 3 | NL | [30] | c.803C>T (p.Thr268Met)/c.915C>A(p.Tyr305*) | MS/NS | KD**KDº | E3 HM/E4 AM | x | A | |||||||||||||||||||||
| 4 | NL | [30] | c.803C>T (p.Thr268Met)/c.915C>A(p.Tyr305*) | MS/NS | KD**KDº | E3 HM/E4 AM | x | A | |||||||||||||||||||||
| 5 | NL | [31] | c.1135G>A(p.Gly379Arg) | MS | KD*KD* | E6 HM | x | x | x | x | x | A | |||||||||||||||||
| 6 | NL | [31] | c.784 + 5 G>C | Spe | KD**KD** | Int2 HM (sk E2) | x | A | |||||||||||||||||||||
| 7 | NL | [31] | c.784 + 5 G>C | Spe | KD**KD** | Int2 HM (sk E2) | x | x | x | x | A | ||||||||||||||||||
| 8 | NL | [31] | c.784 + 5 G>C | Spe | KD**KD** | Int2 HM (sk E2) | x | x | x | x | A | ||||||||||||||||||
| 9 | NL | [31] | c.1487C>T(p.P496L) | MS | KD*KD* | E9 HM | x | A | |||||||||||||||||||||
| 10 | NL | [32] | c.676T>A(p.Trp226Arg) | MS | KD**KD** | E2 HM | x | x | x | A | |||||||||||||||||||
| 11 | NL | [32] | c.676T>A(p.Trp226Arg) | MS | KD**KD** | E2 HM | x | x | A | ||||||||||||||||||||
| 12 | NL | [18] | ND | - | - | - | r | nn | |||||||||||||||||||||
| 13 | NL | [33] | c.1219T>G(p.Tyr407Gly) | MS | KD*KD* | E6 HM | x | a | |||||||||||||||||||||
| 14 | NL | [2] | c.1228 T>A(p.Ser410Thr) | MS | KD*KD* | E6 HM | x | x | x | x | r | r | r | n | n | n | |||||||||||||
| 15 | NL | [34] | c.1351G>A(p.Asp451Asn) | MS | KD*KD* | E7 HM | x | x | x | x | a | ||||||||||||||||||
| 16 | NL | [34] | c.1351G>A(p.Asp451Asn) | MS | KD*KD* | E7 HM | X | A | |||||||||||||||||||||
| 17 | NL | [34] | c.496G>T(p.E166X) | NS | KDºKDº | E1 AM | x | A | |||||||||||||||||||||
| 18 | NL | [35] | c.1645C>T (p.Arg549Trp)/c.863 + 5 G>C | MS/Spe | KD*KD** | E10 HM/Int3 HM | X | x | x | r | r | n | n | ||||||||||||||||
| 19 | NL | [28] | c.1680C>A(p.Cys560Ter) | MS | KD*KD* | E10 HM | X | x | x | a | |||||||||||||||||||
| 20 | NL | [36] | c.1094G>T (Gly365Val) | MS | KD*KD* | E5 HM | X | A | |||||||||||||||||||||
| 21 | NL | [36] | c.1094G>T (Gly365Val) | MS | KD*KD* | E5 HM | x | A | |||||||||||||||||||||
| TOTAL | KDºKDº=1 KD*KD*=10KD**KD**= 6KD**KDº= 2KD*KD**=1 | 11 | 11 | 6 | 10 | 5 | 5 | 1 | 1 | 2 | 2 | 2 | 0 | 1 | 1 | 0 | 0 | 2 | 1 | 0 | 2 | 0 | 1 | 18 |
| Gene | Cerebral Cortex (nTPM Values) | Gene | White Matter (nTPM Values) | Gene | Cerebellum (nTPM Values) | |
|---|---|---|---|---|---|---|
| 1 | CLU | 8112.5 | CLU | 9359.1 | CLU | 7083.7 |
| 2 | APOE | 951 | SPP1 | 1911 | SPP1 | 752 |
| 3 | SPP1 | 743 | TF | 812 | APOE | 680 |
| 4 | EEF1G | 727.9 | LGALS1 | 792 | EEF1G | 623.2 |
| 5 | LGALS1 | 701 | APOE | 763 | LGALS1 | 566 |
| 6 | APP | 548 | EEF1G | 656.7 | TF | 536 |
| 7 | ATP1A2 | 416.2 | APP | 605 | APP | 475 |
| 8 | TIMP1 | 332 | ATP1A2 | 297.4 | ATP1A2 | 409.8 |
| 9 | HSPD1 | 314.1 | CALR | 295.5 | CALR | 242.7 |
| 10 | TSC22D1 | 298.2 | HSP90B1 | 254.2 | EPHX1 | 217 |
| 11 | ATP1A1 | 278.3 | ATP1A1 | 251.1 | TSC22D1 | 214.5 |
| 12 | AHSA1 | 274.8 | EPHX1 | 247.2 | HSPD1 | 209.2 |
| 13 | FKBP4 | 269.2 | HSPD1 | 243.6 | ATP1A1 | 207.4 |
| 14 | STIP1 | 263.1 | TSC22D1 | 242.3 | SQSTM1 | 190.4 |
| 15 | EPHX1 | 251.2 | SQSTM1 | 241.3 | HSP90B1 | 189.8 |
| 16 | SERPINH1 | 238.7 | SCG2 | 230 | APLP2 | 169 |
| 17 | TF | 235 | CHGB | 218 | SPOCK2 | 156 |
| 18 | PLD3 | 231.6 | STIP1 | 204.1 | MAP2K2 | 154 |
| 19 | CALR | 214.2 | HLA-A | 198.2 | AHSA1 | 150.9 |
| 20 | HSP90B1 | 205.9 | PICALM | 196.9 | HADHB | 145.4 |
| 21 | SQSTM1 | 194.2 | SPOCK2 | 196 | CHGB | 137 |
| 22 | MAP2K2 | 192.2 | FKBP4 | 187.2 | FKBP4 | 133 |
| 23 | SCG2 | 161 | AHSA1 | 183.1 | STIP1 | 127.7 |
| 24 | APLP2 | 159 | PLD3 | 182.7 | PICALM | 116.5 |
| 25 | SPOCK2 | 140 | HADHB | 153.1 | TIMP1 | 101 |
| 26 | HADHB | 139.4 | APLP2 | 151 | SERPINH1 | 52.1 |
| 27 | PICALM | 116.4 | MAP2K2 | 146.5 | PLD3 | 31.7 |
| 28 | CHGB | 103 | TIMP1 | 141 | SCG2 | 21.6 |
| 29 | HLA-A | 36.3 | SERPINH1 | 114.4 | HLA-A | 20.1 |
| No. | Description | p-Value | FDR p-Value | Involved Genes |
|---|---|---|---|---|
| 1 | Protein-containing-complex remodeling | 1.97 × 10−6 | 8.49 × 10−3 | APOE, APOA2, ALB |
| 2 | Protein–lipid-complex remodeling | 1.97 × 10−6 | 4.24 × 10−3 | APOE, APOA2, ALB |
| 3 | Plasma-lipoprotein-particle remodeling | 1.97 × 10−6 | 2.83 × 10−3 | APOE, APOA2, ALB |
| 4 | Protein–lipid-complex subunit organization | 2.30 × 10−5 | 2.48 × 10−2 | APOE, APOA2, ALB |
| 5 | Plasma-lipoprotein-particle organization | 2.30 × 10−5 | 1.98 × 10−2 | APOE, APOA2, ALB |
| 6 | Triglyceride-rich lipoprotein-particle remodeling | 2.80 × 10−5 | 2.01 × 10−2 | APOE, APOA2 |
| 7 | Cholesterol efflux | 2.80 × 10−5 | 1.72 × 10−2 | APOE, APOA2 |
| 8 | Lipoprotein metabolic process | 1.08 × 10−4 | 5.81 × 10−2 | APOE, APOA2, APOL1 |
| 9 | Transport | 1.87 × 10−4 | 8.95 × 10−2 | APOA2, COPG1, ARFGAP2, CANX, AP3M1, APLP2, ATP1A1, ATP1A2, CASQ2, ARFGAP1, CP, COPB2, ADAM10, BST2, ANO8, AUP1, AFP, CLU, CKAP4, PPP3CA, ARCN1, CAT, AP3B1, ASHG, APOL1, ASPH, CAND1, CALR, C3, APOE, COPB1, AKAP8, COPA, APP, ALB |
| 10 | Plasma-lipoprotein-particle assembly | 2.22 × 10−4 | 9.57 × 10−2 | APOE, APOA2 |
| 11 | Cholesterol transport | 2.22 × 10−4 | 8.70 × 10−2 | APOE, APOA2 |
| 12 | Organic-hydroxy-compound transport | 2.22 × 10−4 | 7.98 × 10−2 | APOE, APOA2 |
| 13 | Sterol transport | 2.22 × 10−4 | 7.36 × 10−2 | APOE, APOA2 |
| 14 | Sterol homeostasis | 2.22 × 10−4 | 6.84 × 10−2 | APOE, APOA2 |
| 15 | Protein–lipid-complex assembly | 2.22 × 10−4 | 6.38 × 10−2 | APOE, APOA2 |
| 16 | Cholesterol homeostasis | 2.22 × 10−4 | 5.98 × 10−2 | APOE, APOA2 |
| 17 | Establishment of localization | 3.22 × 10−4 | 8.18 × 10−2 | APOA2, COPG1, ARFGAP2, CANX, AP3M1, APLP2, ATP1A1, ATP1A2, CASQ2, ARFGAP1, COPB2, CP, ADAM10, BST2, ANO8, AUP1, CLU, CKAP4, PP3CA, ARCN1, CAT, AP3B1, AHSG, APOL1, ASPH, CAND1, CALR1, C3, APOE, AKAP8, COPB1, COPA, APP, ALB |
| 18 | Lipid transport | 3.42 × 10−4 | 8.21 × 10−2 | APOE, APOA2, APOL1 |
| 19 | Import into cell | 3.73 × 10−4 | 8.47 × 10−2 | CALR, APOE, APP, ALB, CANX, AP3M1, ATP1A1, ATP1A2, APOL1, ASHG |
| 20 | Positive regulation of cholesterol esterification | 5.11 × 10−4 | 1.10 × 10−1 | APOE, APOA2, |
| 21 | Regulation of cholesterol esterification | 5.11 × 10−4 | 1.05 × 10−1 | APOE, APOA2, |
| 31 | Axo-dendritic transport | 6.16 × 10−4 | 8.58 × 10−2 | APP, AP3B1, AP3M1 |
| N | Description | p-Value | FDR q-Value |
|---|---|---|---|
| 1 | Lipid binding | 4.33 × 10−5 | 3.52 × 10−2 |
| 2 | Protein-dimerization activity | 4.56 × 10−5 | 1.85 × 10−2 |
| 3 | Intermembrane cholesterol-transfer activity | 5.11 × 10−4 | 1.38 × 10−1 |
| 4 | Intermembrane sterol-transfer activity | 5.11 × 10−4 | 1.04 × 10−1 |
| 5 | Intermembrane lipid-transfer activity | 5.11 × 10−4 | 8.30 × 10−2 |
| 6 | Sterol-transporter activity | 5.11 × 10−4 | 6.92 × 10−2 |
| 7 | Phosphatidylcholine-sterol O-acyltransferase-activator activity | 5.11 × 10−4 | 5.93 × 10−2 |
| 8 | Cholesterol-transporter activity | 5.11 × 10−4 | 5.19 × 10−2 |
| 9 | Protein kinase A regulatory-subunit binding | 8.02 × 10−4 | 7.24 × 10−2 |
| N | Description | p-Value | FDR q-Value |
|---|---|---|---|
| 1 | Very-low-density lipoprotein particle | 2.68 × 10−6 | 1.47 × 10−3 |
| 2 | Triglyceride-rich plasma-lipoprotein particle | 2.68 × 10−6 | 7.35 × 10−4 |
| 3 | Lipoprotein particle | 2.73 × 10−5 | 4.99 × 10−3 |
| 4 | Plasma-lipoprotein particle | 2.73 × 10−5 | 3.74 × 10−3 |
| 5 | High-density lipoprotein particle | 2.73 × 10−5 | 2.99 × 10−3 |
| 6 | Protein–lipid complex | 2.73 × 10−5 | 2.49 × 10−3 |
| 7 | Chylomicron | 2.80 × 10−5 | 2.19 × 10−3 |
| 8 | Neuron part | 1.40 × 10−4 | 9.56 × 10−3 |
| 9 | Vesicle membrane | 8.45 × 10−4 | 5.14 × 10−2 |
| 10 | Cytoplasmic-vesicle membrane | 8.45 × 10−4 | 4.63 × 10−2 |
| 11 | Cytoplasm | 9.77 × 10−4 | 4.87 × 10−2 |
| N | Pathway Category (Panther ID) | Number of Genes | % of Involved Genes | % of Involved Pathways |
|---|---|---|---|---|
| 1 | Apoptosis-signaling pathway (P00006) | 2 | 0.70% | 2.30% |
| 2 | Interleukin-signaling pathway (P00036) | 1 | 0.40% | 1.20% |
| 3 | Angiogenesis (P00005) | 3 | 1.10% | 3.50% |
| 4 | Alzheimer’s-disease presenilin pathway (P00004) | 3 | 1.10% | 3.50% |
| 5 | Integrin-signalling pathway (P00034) | 8 | 3.00% | 9.30% |
| 6 | Alzheimer’s-disease amyloid-secretase pathway (P00003) | 3 | 1.10% | 3.50% |
| 7 | Opioid-proenkephalin pathway (P05915) | 1 | 0.40% | 1.20% |
| 8 | Insulin/IGF pathway mitogen-activated protein kinase Kinase/MAP-kinase cascade (P00032) | 2 | 0.70% | 2.30% |
| 9 | Inflammation mediated by chemokine- and cytokine-signaling pathway (P00031) | 1 | 0.40% | 1.20% |
| 10 | Enkephalin release (P05913) | 1 | 0.40% | 1.20% |
| 11 | Angiotensin II-stimulated signaling through G proteins and beta-arrestin (P05911) | 2 | 0.70% | 2.30% |
| 12 | Endothelin-signaling pathway (P00019) | 2 | 0.70% | 2.30% |
| 13 | Parkinson’s disease (P00049) | 4 | 1.50% | 4.70% |
| 14 | EGF-receptor-signaling pathway (P00018) | 3 | 1.10% | 3.50% |
| 15 | Gonadotropin-releasing hormone-receptor pathway (P06664) | 6 | 2.20% | 7.00% |
| 16 | Vitamin D metabolism and pathway (P04396) | 1 | 0.40% | 1.20% |
| 17 | PDGF-signaling pathway (P00047) | 2 | 0.70% | 2.30% |
| 18 | Ras pathway (P04393) | 2 | 0.70% | 2.30% |
| 19 | Notch-signaling pathway (P00045) | 2 | 0.70% | 2.30% |
| 20 | Cholesterol biosynthesis (P00014) | 1 | 0.40% | 1.20% |
| 21 | Cadherin-signaling pathway (P00012) | 5 | 1.90% | 5.80% |
| 22 | Blood coagulation (P00011) | 5 | 1.90% | 5.80% |
| 23 | B-cell activation (P00010) | 2 | 0.70% | 2.30% |
| 24 | CCKR-signaling map (P06959) | 5 | 1.90% | 5.80% |
| 25 | Salvage pyrimidine deoxyribonucleotides (P02774) | 1 | 0.40% | 1.20% |
| 26 | p53 pathway (P00059) | 1 | 0.40% | 1.20% |
| 27 | Wnt-signaling pathway (P00057) | 3 | 1.10% | 3.50% |
| 28 | VEGF-signaling pathway (P00056) | 2 | 0.70% | 2.30% |
| 29 | 5-Hydroxytryptamine degredation (P04372) | 1 | 0.40% | 1.20% |
| 30 | Toll-receptor-signaling pathway (P00054) | 3 | 1.10% | 3.50% |
| 31 | T-cell activation (P00053) | 3 | 1.10% | 3.50% |
| 32 | TGF-beta-signaling pathway (P00052) | 2 | 0.70% | 2.30% |
| 33 | FGF-signaling pathway (P00021) | 2 | 0.70% | 2.30% |
| 34 | Arginine biosynthesis (P02728) | 1 | 0.40% | 1.20% |
| Gene | Disease | Disease Class | Number of Associated Genes | GDA Score | EL Score | EI Score |
|---|---|---|---|---|---|---|
| HSPD1 | Spastic paraplegia 13, autosomal dominant | Congenital, hereditary, and neonatal diseases and abnormalities; nervous-system diseases | 1 | 0.92 | 1 | |
| SERPINH1 | Osteogenesis imperfecta Type X | 1 | 0.91 | strong | 1 | |
| TF | Congenital atransferrinemia | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases | 3 | 0.91 | strong | 1 |
| APP | Alzheimer’s disease | Nervous-system diseases; mental disorders | 3397 | 0.9 | 0.981 | |
| APOE | Hyperlipoproteinemia type III | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases | 28 | 0.8 | strong | 0.977 |
| APOE | Alzheimer’s disease 2 | Nervous-system diseases; mental disorders | 16 | 0.8 | strong | 0.96 |
| APOE | Lipoprotein glomerulopathy | Female urogenital diseases and pregnancy complications; male urogenital diseases | 5 | 0.8 | strong | 0.95 |
| ATP1A2 | Migraine, familial hemiplegic 2 | Nervous-system diseases | 6 | 0.8 | 1 | |
| CALR | Primary myelofibrosis | Hemic and lymphatic diseases | 282 | 0.8 | limited | 0.929 |
| MAP2K2 | Cardio-faciocutaneous syndrome | Congenital, hereditary, and neonatal diseases and abnormalities; skin and connective-tissue diseases; cardiovascular diseases | 28 | 0.8 | definitive | 0.966 |
| HADHB | Trifunctional-protein deficiency with myopathy and neuropathy | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases; musculoskeletal diseases; nervous-system diseases; cardiovascular diseases | 23 | 0.78 | definitive | 1 |
| HSPD1 | Leukodystrophy, hypomyelinating 4 | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases; nervous-system diseases | 3 | 0.73 | 1 | |
| APOE | Alzheimer’s disease | Nervous-system diseases; mental disorders | 3397 | 0.7 | 0.946 | |
| APOE | Coronary heart disease | Cardiovascular diseases | 1576 | 0.7 | 0.966 | |
| APOE | Hypercholesterolemia | Nutritional and metabolic diseases | 489 | 0.7 | 0.957 | |
| APOE | Hypertensive disease | Cardiovascular diseases | 2322 | 0.7 | 0.897 | |
| APOE | Sea-blue histiocyte syndrome | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases; nervous-system diseases; hemic and lymphatic diseases | 5 | 0.7 | strong | 1 |
| APP | Dementia | Nervous-system diseases; mental disorders | 816 | 0.7 | strong | 0.964 |
| APP | Cerebral amyloid angiopathy, app-related | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases; nervous-system diseases; cardiovascular diseases | 1 | 0.7 | 1 | |
| ATP1A1 | Charcot–Marie–Tooth disease, axonal, type 2dd | 2 | 0.7 | strong | 1 | |
| SQSTM1 | Amyotrophic lateral sclerosis | Nutritional and metabolic diseases; nervous-system diseases | 1114 | 0.7 | 0.974 | |
| MAP2K2 | Melanoma | Neoplasms | 3087 | 0.67 | 1 | |
| MAP2K2 | Noonan syndrome | Congenital, hereditary, and neonatal diseases and abnormalities; skin and connective-tissue diseases; musculoskeletal diseases; cardiovascular diseases | 85 | 0.64 | limited | 1 |
| ATP1A2 | Alternating hemiplegia of childhood 1 | 2 | 0.61 | 1 | ||
| SQSTM1 | Nonaka myopathy | Congenital, hereditary, and neonatal diseases and abnormalities; musculoskeletal diseases; nervous-system diseases | 132 | 0.61 | strong | 1 |
| APOE | Hyperlipidemia | Nutritional and metabolic diseases | 472 | 0.6 | 0.957 | |
| APOE | Schizophrenia | Mental disorders | 2872 | 0.6 | 0.78 | |
| APOE | Type IIa hyperlipoproteinemia | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases | 201 | 0.6 | 0.889 | |
| APP | Impaired cognition | Mental disorders | 1630 | 0.6 | 0.98 | |
| APP | Cerebral hemorrhage with amyloidosis, hereditary, Dutch type | Congenital, hereditary, and neonatal diseases and abnormalities; nutritional and metabolic diseases; nervous-system diseases; cardiovascular diseases | 20 | 0.6 | 1 | |
| CALR | Myelofibrosis | neoplasms; hemic and lymphatic diseases | 163 | 0.6 | limited | 0.933 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma-Lara, I.; García Alonso-Themann, P.; Pérez-Durán, J.; Godínez-Aguilar, R.; Bonilla-Delgado, J.; Gómez-Archila, D.; Espinosa-García, A.M.; Nolasco-Quiroga, M.; Victoria-Acosta, G.; López-Ornelas, A.; et al. Potential Role of Protein Kinase FAM20C on the Brain in Raine Syndrome, an In Silico Analysis. Int. J. Mol. Sci. 2023, 24, 8904. https://doi.org/10.3390/ijms24108904
Palma-Lara I, García Alonso-Themann P, Pérez-Durán J, Godínez-Aguilar R, Bonilla-Delgado J, Gómez-Archila D, Espinosa-García AM, Nolasco-Quiroga M, Victoria-Acosta G, López-Ornelas A, et al. Potential Role of Protein Kinase FAM20C on the Brain in Raine Syndrome, an In Silico Analysis. International Journal of Molecular Sciences. 2023; 24(10):8904. https://doi.org/10.3390/ijms24108904
Chicago/Turabian StylePalma-Lara, Icela, Patricia García Alonso-Themann, Javier Pérez-Durán, Ricardo Godínez-Aguilar, José Bonilla-Delgado, Damián Gómez-Archila, Ana María Espinosa-García, Manuel Nolasco-Quiroga, Georgina Victoria-Acosta, Adolfo López-Ornelas, and et al. 2023. "Potential Role of Protein Kinase FAM20C on the Brain in Raine Syndrome, an In Silico Analysis" International Journal of Molecular Sciences 24, no. 10: 8904. https://doi.org/10.3390/ijms24108904
APA StylePalma-Lara, I., García Alonso-Themann, P., Pérez-Durán, J., Godínez-Aguilar, R., Bonilla-Delgado, J., Gómez-Archila, D., Espinosa-García, A. M., Nolasco-Quiroga, M., Victoria-Acosta, G., López-Ornelas, A., Serrano-Bello, J. C., Olguín-García, M. G., & Palacios-Reyes, C. (2023). Potential Role of Protein Kinase FAM20C on the Brain in Raine Syndrome, an In Silico Analysis. International Journal of Molecular Sciences, 24(10), 8904. https://doi.org/10.3390/ijms24108904