
Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy

 , , and
, , and
Abstract
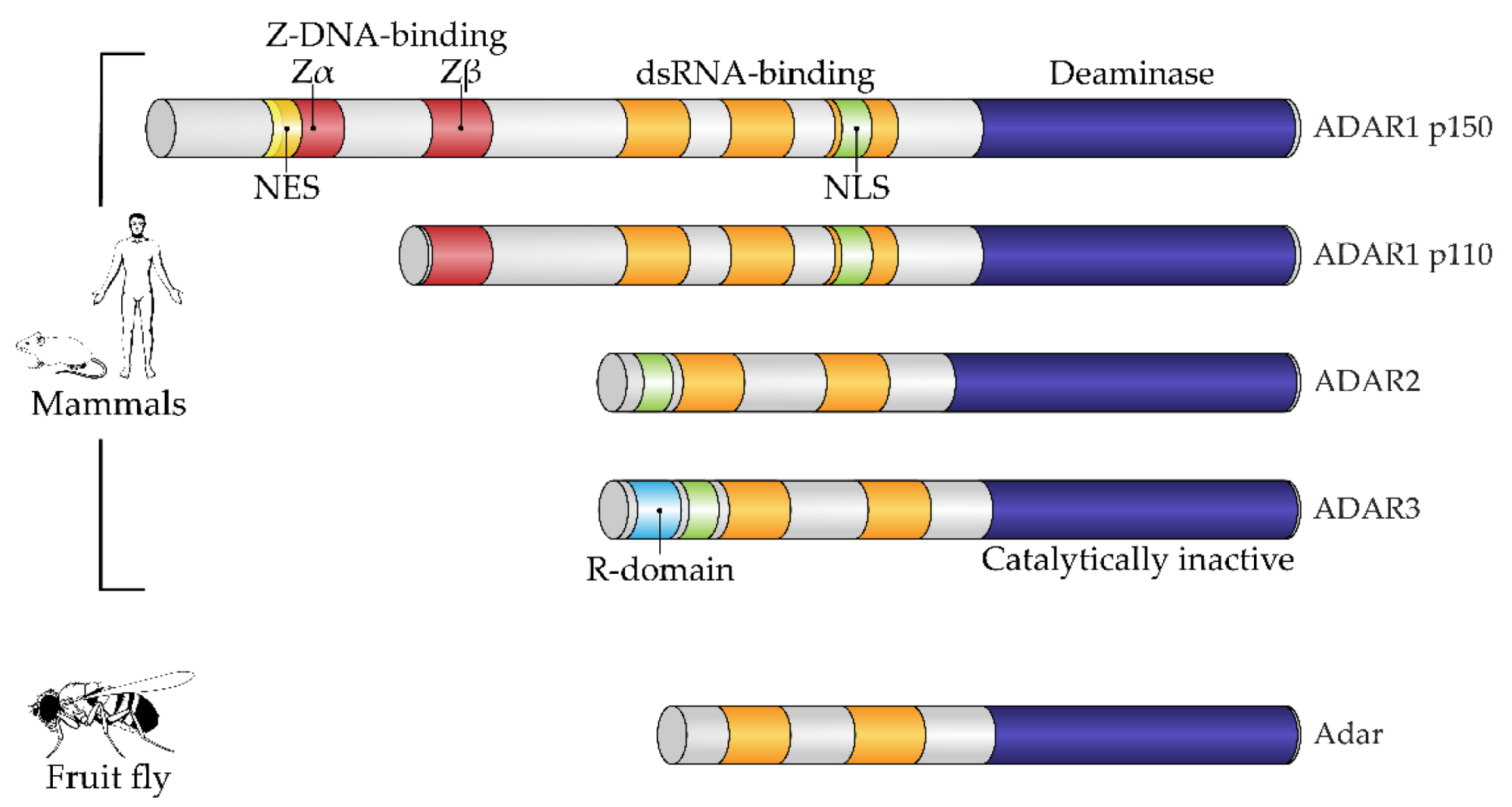
1. Introduction: Adenosine-to-Inosine RNA Editing by ADAR Enzymes
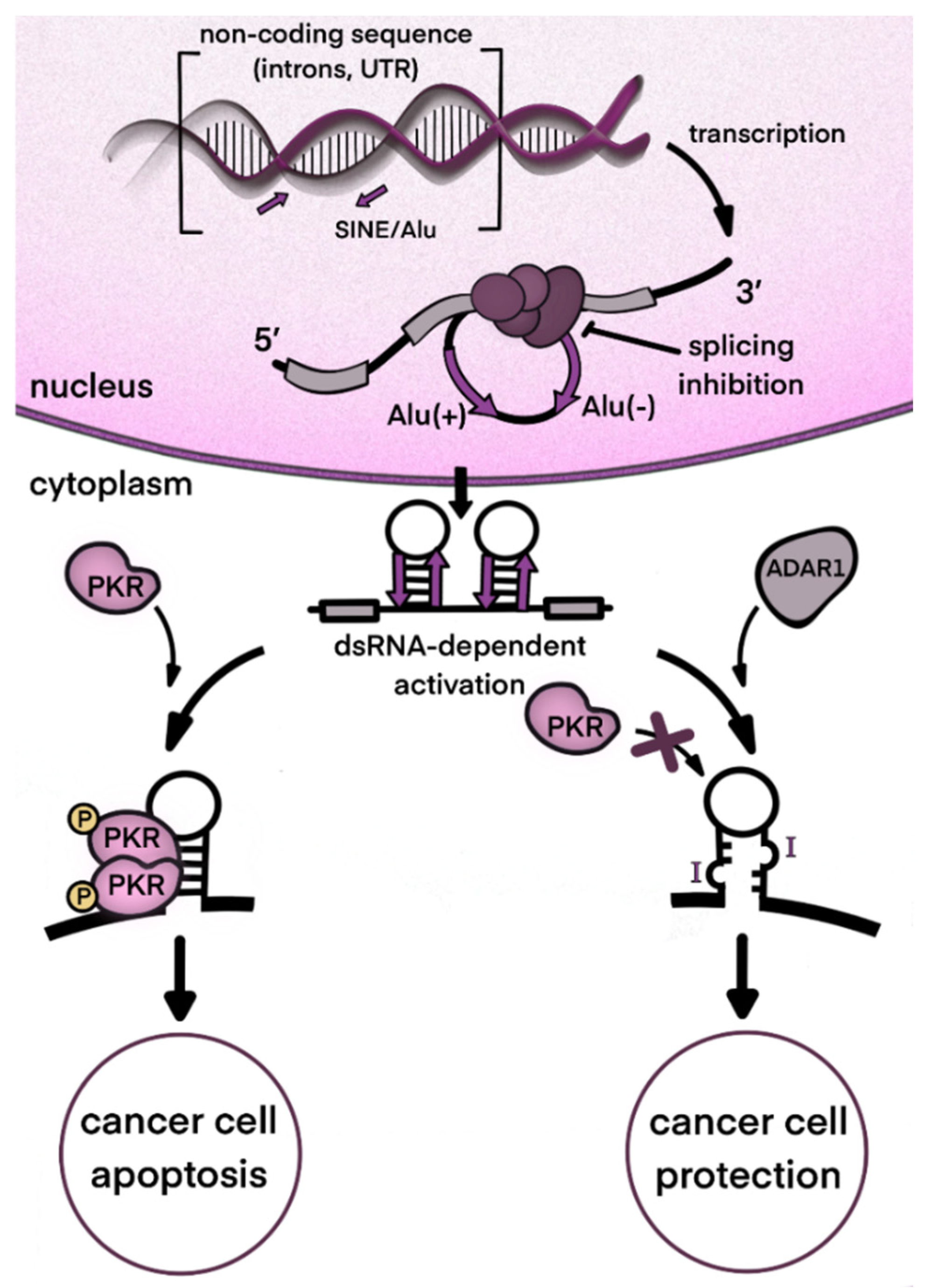
2. ADAR1 and Type I Interferon Signaling in Cancer
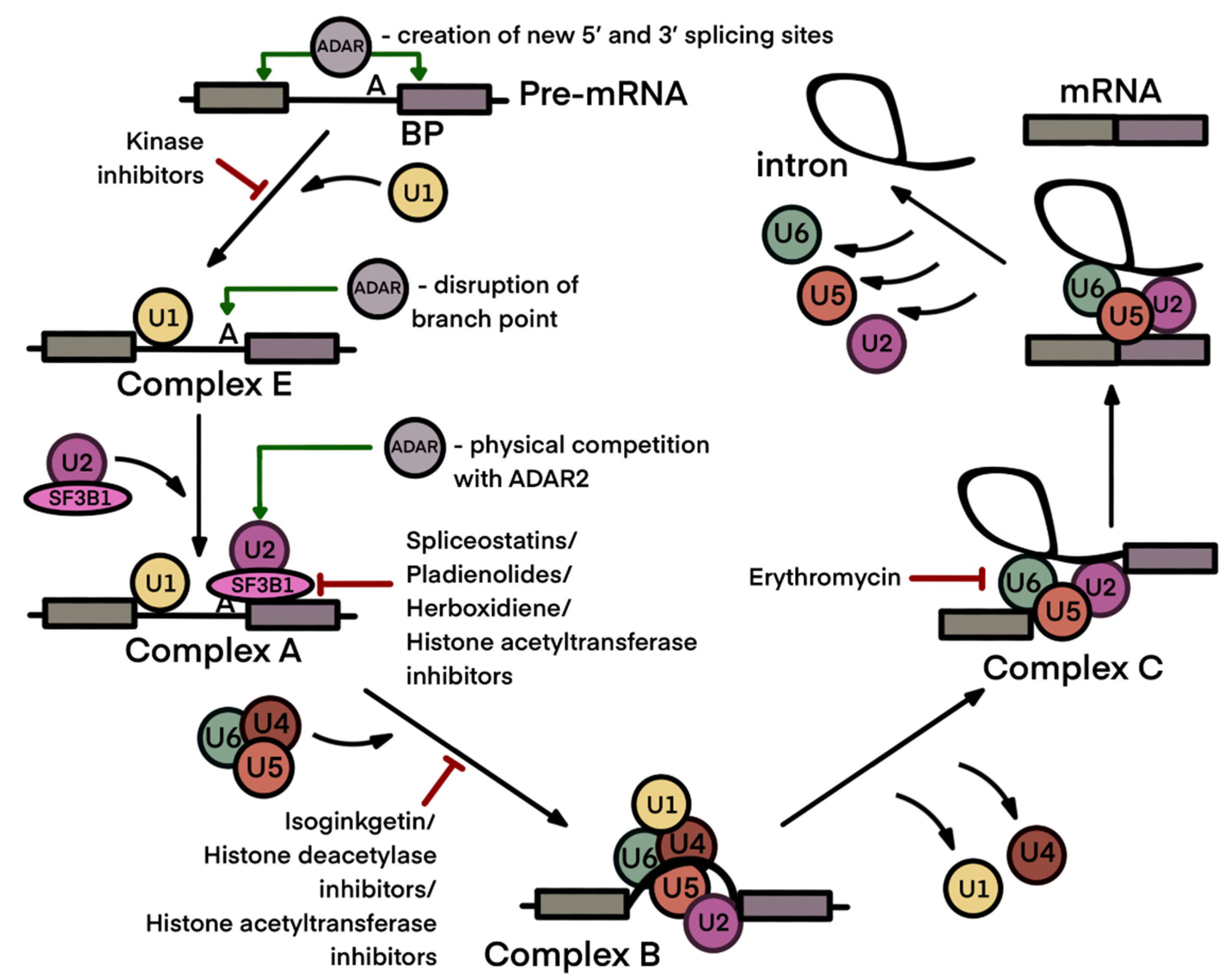
3. Interdependence of A-to-I Editing and Splicing of mRNA
4. Anticancer Therapeutics Targeting the Spliceosome Machinery

5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bass, B.L.; Weintraub, H. An Unwinding Activity That Covalently Modifies Its Double-Stranded RNA Substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Duan, Y.; Tang, X.; Lu, J. Evolutionary Driving Forces of A-to-I Editing in Metazoans. Wiley Interdiscip. Rev. RNA 2022, 13, e1666. [Google Scholar] [CrossRef] [PubMed]
- Luciano, D.J.; Mirsky, H.; Vendetti, N.J.; Maas, S. RNA Editing of a miRNA Precursor. RNA 2004, 10, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.H.; Castro, M.M.; Aboul-ela, F.; Tinoco, I., Jr. Base Pairing Involving Deoxyinosine: Implications for Probe Design. Nucleic Acids Res. 1985, 13, 8927–8938. [Google Scholar] [CrossRef]
- Basilio, C.; Wahba, A.J.; Lengyel, P.; Speyer, J.F.; Ochoa, S. Synthetic Polynucleotides and the Amino Acid Code. V. Proc. Natl. Acad. Sci. USA 1962, 48, 613–616. [Google Scholar] [CrossRef]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA Editing in Brain Controls a Determinant of Ion Flow in Glutamate-Gated Channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. A Developmentally Regulated Activity That Unwinds RNA Duplexes. Cell 1987, 48, 607–613. [Google Scholar] [CrossRef]
- Grice, L.F.; Degnan, B.M. The Origin of the ADAR Gene Family and Animal RNA Editing. BMC Evol. Biol. 2015, 15, 4. [Google Scholar] [CrossRef]
- Yablonovitch, A.L.; Deng, P.; Jacobson, D.; Li, J.B. The Evolution and Adaptation of A-to-I RNA Editing. PLoS Genet. 2017, 13, e1007064. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and Regulation by Interferon of a Double-Stranded-RNA-Specific Adenosine Deaminase from Human Cells: Evidence for Two Forms of the Deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef]
- Wulff, B.-E.; Nishikura, K. Substitutional A-to-I RNA Editing. Wiley Interdiscip. Rev. RNA 2010, 1, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.M.; Chu, H.; Rueter, S.M.; Hutchinson, L.K.; Canton, H.; Sanders-Bush, E.; Emeson, R.B. Regulation of Serotonin-2C Receptor G-Protein Coupling by RNA Editing. Nature 1997, 387, 303–308. [Google Scholar] [CrossRef]
- Kliuchnikova, A.A.; Goncharov, A.O.; Levitsky, L.I.; Pyatnitskiy, M.A.; Novikova, S.E.; Kuznetsova, K.G.; Ivanov, M.V.; Ilina, I.Y.; Farafonova, T.E.; Zgoda, V.G.; et al. Proteome-Wide Analysis of ADAR-Mediated Messenger RNA Editing during Fruit Fly Ontogeny. J. Proteome Res. 2020, 19, 4046–4060. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.J.; Keegan, L.P.; O’Connell, M.A.; Reenan, R.A. dADAR, a Drosophila Double-Stranded RNA-Specific Adenosine Deaminase Is Highly Developmentally Regulated and Is Itself a Target for RNA Editing. RNA 2000, 6, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR Protein Family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Liscovitch-Brauer, N.; Alon, S.; Porath, H.T.; Elstein, B.; Unger, R.; Ziv, T.; Admon, A.; Levanon, E.Y.; Rosenthal, J.J.C.; Eisenberg, E. Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 2017, 169, 191–202.e11. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Tackett, M.R.; Nechkin, S.; Shtokalo, D.; Antonets, D.; Savva, Y.A.; Maloney, R.; Kapranov, P.; Lawrence, C.E.; Reenan, R.A. Genome-Wide Analysis of A-to-I RNA Editing by Single-Molecule Sequencing in Drosophila. Nat. Struct. Mol. Biol. 2013, 20, 1333–1339. [Google Scholar] [CrossRef]
- Xuan, N.; Bu, X.; Liu, Y.Y.; Yang, X.; Liu, G.X.; Fan, Z.X.; Bi, Y.P.; Yang, L.Q.; Lou, Q.N.; Rajashekar, B.; et al. Molecular Evidence of RNA Editing in Bombyx Chemosensory Protein Family. PLoS ONE 2014, 9, e86932. [Google Scholar] [CrossRef]
- Jepson, J.E.C.; Reenan, R.A. Adenosine-to-Inosine Genetic Recoding Is Required in the Adult Stage Nervous System for Coordinated Behavior in Drosophila. J. Biol. Chem. 2009, 284, 31391–31400. [Google Scholar] [CrossRef]
- Garrett, S.; Rosenthal, J.J.C. RNA Editing Underlies Temperature Adaptation in K+ Channels from Polar Octopuses. Science 2012, 335, 848–851. [Google Scholar] [CrossRef]
- Yablonovitch, A.L.; Fu, J.; Li, K.; Mahato, S.; Kang, L.; Rashkovetsky, E.; Korol, A.B.; Tang, H.; Michalak, P.; Zelhof, A.C.; et al. Regulation of Gene Expression and RNA Editing in Drosophila Adapting to Divergent Microclimates. Nat. Commun. 2017, 8, 1570. [Google Scholar] [CrossRef] [PubMed]
- Picimbon, J.F. RNA Mutations: Source of Life. Gene Technol. 2014, 4, 112. [Google Scholar] [CrossRef]
- Jiang, D.; Zhang, J. Author Correction: The Preponderance of Nonsynonymous A-to-I RNA Editing in Coleoids Is Nonadaptive. Nat. Commun. 2021, 12, 4200. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, W.; Li, Q. Origins and Evolution of ADAR-Mediated RNA Editing. IUBMB Life 2009, 61, 572–578. [Google Scholar] [CrossRef]
- Goncharov, A.O.; Kliuchnikova, A.A.; Nasaev, S.S.; Moshkovskii, S.A. RNA Editing by ADAR Adenosine Deaminases: From Molecular Plasticity of Neural Proteins to the Mechanisms of Human Cancer. Biochem. (Mosc.) 2019, 84, 896–904. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, J. Human Coding RNA Editing Is Generally Nonadaptive. Proc. Natl. Acad. Sci. USA 2014, 111, 3769–3774. [Google Scholar] [CrossRef]
- Kuznetsova, K.G.; Kliuchnikova, A.A.; Ilina, I.U.; Chernobrovkin, A.L.; Novikova, S.E.; Farafonova, T.E.; Karpov, D.S.; Ivanov, M.V.; Goncharov, A.O.; Ilgisonis, E.V.; et al. Proteogenomics of Adenosine-to-Inosine RNA Editing in the Fruit Fly. J. Proteome Res. 2018, 17, 3889–3903. [Google Scholar] [CrossRef]
- Levitsky, L.I.; Kliuchnikova, A.A.; Kuznetsova, K.G.; Karpov, D.S.; Ivanov, M.V.; Pyatnitskiy, M.A.; Kalinina, O.V.; Gorshkov, M.V.; Moshkovskii, S.A. Adenosine-to-Inosine RNA Editing in Mouse and Human Brain Proteomes. Proteomics 2019, 19, e1900195. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.A.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 Cause Aicardi-Goutières Syndrome Associated with a Type I Interferon Signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef] [PubMed]
- Bajad, P.; Ebner, F.; Amman, F.; Szabó, B.; Kapoor, U.; Manjali, G.; Hildebrandt, A.; Janisiw, M.P.; Jantsch, M.F. An Internal Deletion of ADAR Rescued by MAVS Deficiency Leads to a Minute Phenotype. Nucleic Acids Res. 2020, 48, 3286–3303. [Google Scholar] [CrossRef] [PubMed]
- Brusa, R.; Zimmermann, F.; Koh, D.S.; Feldmeyer, D.; Gass, P.; Seeburg, P.H.; Sprengel, R. Early-Onset Epilepsy and Postnatal Lethality Associated with an Editing-Deficient GluR-B Allele in Mice. Science 1995, 270, 1677–1680. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Y.; Sedmík, J.; Fitzgerald, M.P.; Halevy, R.S.; Keegan, L.P.; Helbig, I.; Basel-Salmon, L.; Cohen, L.; Straussberg, R.; Chung, W.K.; et al. Bi-Allelic ADARB1 Variants Associated with Microcephaly, Intellectual Disability, and Seizures. Am. J. Hum. Genet. 2020, 106, 467–483. [Google Scholar] [CrossRef]
- Seeburg, P.H.; Higuchi, M.; Sprengel, R. RNA Editing of Brain Glutamate Receptor Channels: Mechanism and Physiology. Brain Res. Brain Res. Rev. 1998, 26, 217–229. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point Mutation in an AMPA Receptor Gene Rescues Lethality in Mice Deficient in the RNA-Editing Enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Wang, H.; Chen, S.; Wei, J.; Song, G.; Zhao, Y. A-to-I RNA Editing in Cancer: From Evaluating the Editing Level to Exploring the Editing Effects. Front. Oncol. 2020, 10, 632187. [Google Scholar] [CrossRef]
- Chan, T.H.M.; Qamra, A.; Tan, K.T.; Guo, J.; Yang, H.; Qi, L.; Lin, J.S.; Ng, V.H.E.; Song, Y.; Hong, H.; et al. ADAR-Mediated RNA Editing Predicts Progression and Prognosis of Gastric Cancer. Gastroenterology 2016, 151, 637–650.e10. [Google Scholar] [CrossRef]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered Adenosine-to-Inosine RNA Editing in Human Cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Galore-Haskel, G.; Nemlich, Y.; Greenberg, E.; Ashkenazi, S.; Hakim, M.; Itzhaki, O.; Shoshani, N.; Shapira-Fromer, R.; Ben-Ami, E.; Ofek, E.; et al. A Novel Immune Resistance Mechanism of Melanoma Cells Controlled by the ADAR1 Enzyme. Oncotarget 2015, 6, 28999–29015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.-C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA Editing Derived Epitopes Function as Cancer Antigens to Elicit Immune Responses. Nat. Commun. 2018, 9, 3919. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-B.; Liao, X.-Y.; Zhang, J.-B.; Wang, F.; Qin, H.-D.; Zhang, L.; Shugart, Y.Y.; Zeng, Y.-X.; Jia, W.-H. ADAR2 Functions as a Tumor Suppressor via Editing IGFBP7 in Esophageal Squamous Cell Carcinoma. Int. J. Oncol. 2017, 50, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Prados-Carvajal, R.; Fernández-Ávila, M.J.; Silva, S.; Silvestris, D.A.; Endara-Coll, M.; Rodríguez-Real, G.; Domingo-Prim, J.; Mejías-Navarro, F.; Romero-Franco, A.; et al. ADAR-Mediated RNA Editing of DNA:RNA Hybrids Is Required for DNA Double Strand Break Repair. Nat. Commun. 2021, 12, 5512. [Google Scholar] [CrossRef]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 Is a New Target of METTL3 and Plays a pro-Oncogenic Role in Glioblastoma by an Editing-Independent Mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, J.H.; Park, J.-E.; Cho, J.; Yi, H.; Kim, V.N. PKR Is Activated by Cellular dsRNAs during Mitosis and Acts as a Mitotic Regulator. Genes Dev. 2014, 28, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.-Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810.e13. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. Mda-5: An Interferon-Inducible Putative RNA Helicase with Double-Stranded RNA-Dependent ATPase Activity and Melanoma Growth-Suppressive Properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA Helicase RIG-I Has an Essential Function in Double-Stranded RNA-Induced Innate Antiviral Responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of Genes Differentially Regulated by Interferon Alpha, Beta, or Gamma Using Oligonucleotide Arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Davies, M.V.; Pathak, V.K.; Hershey, J.W. The Phosphorylation State of Eucaryotic Initiation Factor 2 Alters Translational Efficiency of Specific mRNAs. Mol. Cell. Biol. 1989, 9, 946–958. [Google Scholar]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and Their Stimulated Genes in the Tumor Microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Koeris, M.; Funke, L.; Shrestha, J.; Rich, A.; Maas, S. Modulation of ADAR1 Editing Activity by Z-RNA in Vitro. Nucleic Acids Res. 2005, 33, 5362–5370. [Google Scholar] [CrossRef] [PubMed]
- Lavut, A.; Raveh, D. Sequestration of Highly Expressed mRNAs in Cytoplasmic Granules, P-Bodies, and Stress Granules Enhances Cell Viability. PLoS Genet. 2012, 8, e1002527. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.-W.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.-B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of Splicing Catalysis for Therapeutic Targeting of Leukemia with Mutations in Genes Encoding Spliceosomal Proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Gannon, H.S.; Zou, T.; Kiessling, M.K.; Gao, G.F.; Cai, D.; Choi, P.S.; Ivan, A.P.; Buchumenski, I.; Berger, A.C.; Goldstein, J.T.; et al. Identification of ADAR1 Adenosine Deaminase Dependency in a Subset of Cancer Cells. Nat. Commun. 2018, 9, 5450. [Google Scholar] [CrossRef]
- Bhate, A.; Sun, T.; Li, J.B. ADAR1: A New Target for Immuno-Oncology Therapy. Mol. Cell 2019, 73, 866–868. [Google Scholar] [CrossRef]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in Tumours Overcomes Resistance to Immune Checkpoint Blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- Sakurai, M.; Shiromoto, Y.; Ota, H.; Song, C.; Kossenkov, A.V.; Wickramasinghe, J.; Showe, L.C.; Skordalakes, E.; Tang, H.-Y.; Speicher, D.W.; et al. ADAR1 Controls Apoptosis of Stressed Cells by Inhibiting Staufen1-Mediated mRNA Decay. Nat. Struct. Mol. Biol. 2017, 24, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Herzner, A.-M.; Khan, Z.; Van Nostrand, E.L.; Chan, S.; Cuellar, T.; Chen, R.; Pechuan-Jorge, X.; Komuves, L.; Solon, M.; Modrusan, Z.; et al. ADAR and hnRNPC Deficiency Synergize in Activating Endogenous dsRNA-Induced Type I IFN Responses. J. Exp. Med. 2021, 218, e20201833. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golji, J.; Brodeur, L.K.; Chung, F.S.; Chen, J.T.; deBeaumont, R.S.; Bullock, C.P.; Jones, M.D.; Kerr, G.; Li, L.; et al. Tumor-Derived IFN Triggers Chronic Pathway Agonism and Sensitivity to ADAR Loss. Nat. Med. 2019, 25, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.-Y.; Yang, W.-Y.; Zhang, L.-H.; Li, L.; Xie, F.; Li, H.-Y.; Chen, X.-Y.; Tu, Z.; Li, Y.; Chen, Y.; et al. 8-Chloro-Adenosine Inhibits Proliferation of MDA-MB-231 and SK-BR-3 Breast Cancer Cells by Regulating ADAR1/p53 Signaling Pathway. Cell Transplant. 2020, 29, 963689720958656. [Google Scholar] [CrossRef]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.-J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Cottrell, K.A.; Soto-Torres, L.; Dizon, M.G.; Weber, J.D. 8-Azaadenosine and 8-Chloroadenosine Are Not Selective Inhibitors of ADARLack of ADAR Inhibitors. Cancer Res. Commun. 2021, 1, 56–64. [Google Scholar] [CrossRef]
- Hang, P.N.T.; Tohda, M.; Tezuka, Y.; Matsumoto, K. Influence of an Adenosine Deaminase Inhibitor, Erythro-9-(2-Hydroxy-3-Nonyl) Adenine Hydrochloride, on 5-HT2CR mRNA Editing in Primary Cultured Cortical Cells. Biol. Pharm. Bull. 2010, 33, 527–529. [Google Scholar] [CrossRef][Green Version]
- Laurencikiene, J.; Källman, A.M.; Fong, N.; Bentley, D.L.; Ohman, M. RNA Editing and Alternative Splicing: The Importance of Co-Transcriptional Coordination. EMBO Rep. 2006, 7, 303–307. [Google Scholar] [CrossRef]
- Solomon, O.; Oren, S.; Safran, M.; Deshet-Unger, N.; Akiva, P.; Jacob-Hirsch, J.; Cesarkas, K.; Kabesa, R.; Amariglio, N.; Unger, R.; et al. Global Regulation of Alternative Splicing by Adenosine Deaminase Acting on RNA (ADAR). RNA 2013, 19, 591–604. [Google Scholar] [CrossRef]
- Hsiao, Y.-H.E.; Bahn, J.H.; Yang, Y.; Lin, X.; Tran, S.; Yang, E.-W.; Quinones-Valdez, G.; Xiao, X. RNA Editing in Nascent RNA Affects Pre-mRNA Splicing. Genome Res. 2018, 28, 812–823. [Google Scholar] [CrossRef]
- Higuchi, M.; Single, F.N.; Köhler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA Editing of AMPA Receptor Subunit GluR-B: A Base-Paired Intron-Exon Structure Determines Position and Efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Kapoor, U.; Licht, K.; Amman, F.; Jakobi, T.; Martin, D.; Dieterich, C.; Jantsch, M.F. ADAR-Deficiency Perturbs the Global Splicing Landscape in Mouse Tissues. Genome Res. 2020, 30, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Weber, A.; Maly, K.; Manjaly, G.; Deek, J.; Tsvyetkova, O.; Stulić, M.; Toca-Herrera, J.L.; Jantsch, M.F. A-to-I RNA Editing of Filamin A Regulates Cellular Adhesion, Migration and Mechanical Properties. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.J.; Shen, H.; An, O.; Hong, H.; Li, J.; Song, Y.; Han, J.; Tay, D.J.T.; Ng, V.H.E.; Bellido Molias, F.; et al. Cis- and Trans-Regulations of Pre-mRNA Splicing by RNA Editing Enzymes Influence Cancer Development. Nat. Commun. 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Ma, X.; Zeng, F.; Yan, J. RNA Editing Regulates lncRNA Splicing in Human Early Embryo Development. PLoS Comput. Biol. 2021, 17, e1009630. [Google Scholar] [CrossRef]
- Wu, D.; Zang, Y.-Y.; Shi, Y.-Y.; Ye, C.; Cai, W.-M.; Tang, X.-H.; Zhao, L.; Liu, Y.; Gan, Z.; Chen, G.-Q.; et al. Distant Coupling between RNA Editing and Alternative Splicing of the Osmosensitive Cation Channel Tmem63b. J. Biol. Chem. 2020, 295, 18199–18212. [Google Scholar] [CrossRef]
- Tariq, A.; Garncarz, W.; Handl, C.; Balik, A.; Pusch, O.; Jantsch, M.F. RNA-Interacting Proteins Act as Site-Specific Repressors of ADAR2-Mediated RNA Editing and Fluctuate upon Neuronal Stimulation. Nucleic Acids Res. 2013, 41, 2581–2593. [Google Scholar] [CrossRef]
- Shanmugam, R.; Zhang, F.; Srinivasan, H.; Charles Richard, J.L.; Liu, K.I.; Zhang, X.; Woo, C.W.A.; Chua, Z.H.M.; Buschdorf, J.P.; Meaney, M.J.; et al. SRSF9 Selectively Represses ADAR2-Mediated Editing of Brain-Specific Sites in Primates. Nucleic Acids Res. 2018, 46, 7379–7395. [Google Scholar] [CrossRef]
- Huang, H.; Kapeli, K.; Jin, W.; Wong, Y.P.; Arumugam, T.V.; Koh, J.H.; Srimasorn, S.; Mallilankaraman, K.; Chua, J.J.E.; Yeo, G.W.; et al. Tissue-Selective Restriction of RNA Editing of CaV1.3 by Splicing Factor SRSF9. Nucleic Acids Res. 2018, 46, 7323–7338. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The Role of Alternative Splicing in Cancer: From Oncogenesis to Drug Resistance. Drug Resist. Updates 2020, 53, 100728. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Chhipi-Shrestha, J.K.; Yoshida, M. Splicing Modulators: On the Way from Nature to Clinic. J. Antibiot. 2021, 74, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating Splicing with Small Molecular Inhibitors of the Spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8, e1381. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A Targets SF3b and Inhibits Both Splicing and Nuclear Retention of Pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Miura, T.; Kuzuya, K.; Inoue, A.; Won Ki, S.; Horinouchi, S.; Yoshida, T.; Kunoh, T.; Koseki, K.; Mino, K.; et al. Identification of SAP155 as the Target of GEX1A (Herboxidiene), an Antitumor Natural Product. ACS Chem. Biol. 2011, 6, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing Factor SF3b as a Target of the Antitumor Natural Product Pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Tsai, J.H.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B.; et al. Splicing Modulators Act at the Branch Point Adenosine Binding Pocket Defined by the PHF5A-SF3b Complex. Nat. Commun. 2017, 8, 15522. [Google Scholar] [CrossRef]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The Cryo-EM Structure of the SF3b Spliceosome Complex Bound to a Splicing Modulator Reveals a Pre-mRNA Substrate Competitive Mechanism of Action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef]
- Wan, Y.; Zheng, X.; Chen, H.; Guo, Y.; Jiang, H.; He, X.; Zhu, X.; Zheng, Y. Splicing Function of Mitotic Regulators Links R-Loop-Mediated DNA Damage to Tumor Cell Killing. J. Cell Biol. 2015, 209, 235–246. [Google Scholar] [CrossRef]
- Han, C.; Khodadadi-Jamayran, A.; Lorch, A.H.; Jin, Q.; Serafin, V.; Zhu, P.; Politanska, Y.; Sun, L.; Gutierrez-Diaz, B.T.; Pryzhkova, M.V.; et al. SF3B1 Homeostasis Is Critical for Survival and Therapeutic Response in T Cell Leukemia. Sci. Adv. 2022, 8, eabj8357. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, Q.; Liu, X.; Ji, Y.; Chao, H.-P.; Liu, Y.; Tracz, A.; Kirk, J.; Buonamici, S.; Zhu, P.; et al. Intron Retention Is a Hallmark and Spliceosome Represents a Therapeutic Vulnerability in Aggressive Prostate Cancer. Nat. Commun. 2020, 11, 2089. [Google Scholar] [CrossRef]
- Hsu, T.Y.-T.; Simon, L.M.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.V.; Sun, T.; Kurley, S.J.; Tyagi, S.; et al. The Spliceosome Is a Therapeutic Vulnerability in MYC-Driven Cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.K.; Kumar, D.; Villa, R.; La Clair, J.J.; Benner, C.; Sasik, R.; Jones, H.; Ghia, E.M.; Rassenti, L.Z.; Kipps, T.J.; et al. Targeting the Spliceosome in Chronic Lymphocytic Leukemia with the Macrolides FD-895 and Pladienolide-B. Haematologica 2015, 100, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F.; et al. The SF3B1 Inhibitor Spliceostatin A (SSA) Elicits Apoptosis in Chronic Lymphocytic Leukaemia Cells through Downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Bowling, E.A.; Wang, J.H.; Gong, F.; Wu, W.; Neill, N.J.; Kim, I.S.; Tyagi, S.; Orellana, M.; Kurley, S.J.; Dominguez-Vidaña, R.; et al. Spliceosome-Targeted Therapies Trigger an Antiviral Immune Response in Triple-Negative Breast Cancer. Cell 2021, 184, 384–403.e21. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Matlin, A.J.; Lowell, A.M.; Moore, M.J. The Biflavonoid Isoginkgetin Is a General Inhibitor of Pre-mRNA Splicing. J. Biol. Chem. 2008, 283, 33147–33154. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, F.Q.; Merkhofer, E.C.; Johnson, T.L. Dynamic Histone Acetylation Is Critical for Cotranscriptional Spliceosome Assembly and Spliceosomal Rearrangements. Proc. Natl. Acad. Sci. USA 2011, 108, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; van Santen, M.A.; Schwienhorst, A.; Urlaub, H.; Lührmann, R. Stalling of Spliceosome Assembly at Distinct Stages by Small-Molecule Inhibitors of Protein Acetylation and Deacetylation. RNA 2009, 15, 153–175. [Google Scholar] [CrossRef]
- Thapar, R. Structural Basis for Regulation of RNA-Binding Proteins by Phosphorylation. ACS Chem. Biol. 2015, 10, 652–666. [Google Scholar] [CrossRef]
- Hertweck, M.; Hiller, R.; Mueller, M.W. Inhibition of Nuclear Pre-mRNA Splicing by Antibiotics in Vitro. Eur. J. Biochem. 2002, 269, 175–183. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T.; et al. Final Results of a Phase 2, Open-Label Study of Indisulam, Idarubicin, and Cytarabine in Patients with Relapsed or Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef]
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Eckold, C.; Valbuena, G.; Liu, Y.; Kouloura, E.; Poon, E.; da Costa, B.M.; et al. Indisulam Targets RNA Splicing and Metabolism to Serve as a Novel Therapeutic Strategy for High-Risk Neuroblastoma. Nature Comm. 2022, 13, 1380. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Quarni, W.; Goralski, M.; Wan, S.; Jin, H.; Van de Velde, L.-A.; Fang, J.; Wu, Q.; Abu-Zaid, A.; Wang, T.; et al. Targeting the Spliceosome through RBM39 Degradation Results in Exceptional Responses in High-Risk Neuroblastoma Models. Sci. Adv. 2021, 7, eabj5405. [Google Scholar] [CrossRef]
- Eskens, F.A.L.M.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.A.; Mizui, Y.; Wiemer, E.A.C.; Carreras, M.J.; Baselga, J.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A Phase I, Open-Label, Single-Arm, Dose-Escalation Study of E7107, a Precursor Messenger Ribonucleic Acid (pre-mRNA) Splicesome Inhibitor Administered Intravenously on Days 1 and 8 Every 21 Days to Patients with Solid Tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an Orally Available Small-Molecule Splicing Modulator, Induces Lethality in Spliceosome-Mutant Cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A Day in the Life of the Spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Compound | Target/Effect | Clinical Trials |
|---|---|---|---|
| RNA splicing inhibitors | |||
| Spliceostatins | Spliceostatin A | Target SF3B1, block A complex assembly | - |
| Sudemycin D6 | - | ||
| Meayamycin B | - | ||
| Pladienolides | FD-895 | - | |
| Pladienolide B | - | ||
| E7107 | + | ||
| H3B-8800 | + | ||
| Herboxidiene | Herboxidiene | - | |
| GEX | - | ||
| 18-Deoxyherboxidiene | - | ||
| Isoginkgetin | Isoginkgetin | Prevents binding of U4/U5/U6 tri-snRNP to the A complex | - |
| Histone deacetylase (HDAC) inhibitors | Suberoylanilide hydroxamic acid | B complex | - |
| Splitomicin | - | ||
| Dihydrocoumarin | - | ||
| Histone acetyltransferase inhibitors | Garcinol | A complex | - |
| AA | B complex | - | |
| BA3 | - | ||
| Kinase inhibitors | Diospyrin | H/E complex | - |
| Chlorhexidine | - | ||
| TG003 | - | ||
| Antibiotics | Chlortetracycline | Early spliceosome assembly | - |
| Streptomycin | - | ||
| Erythromycin | C complex | - | |
| Sulfanilamide | Indisulam | Degradation of splicing factor RBM39 | + |
| E7820 | - | ||
| CQS | - | ||
| Tasisulam | + | ||
| ADAR RNA-editing inhibitors | |||
| Adenosine analogs | 8-Azaadenosine | ADAR1 | - |
| 8-Chloroadenosine | - | ||
| Adenine analog | Erythro-9-(2-hydroxy-3-nonyl) adenine hydrochloride (EHNA) | ADA, a metabolic adenosine deaminase; EHNA also inhibited some ADAR2 editing events | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goncharov, A.O.; Shender, V.O.; Kuznetsova, K.G.; Kliuchnikova, A.A.; Moshkovskii, S.A. Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 5240. https://doi.org/10.3390/ijms23095240
Goncharov AO, Shender VO, Kuznetsova KG, Kliuchnikova AA, Moshkovskii SA. Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy. International Journal of Molecular Sciences. 2022; 23(9):5240. https://doi.org/10.3390/ijms23095240
Chicago/Turabian StyleGoncharov, Anton O., Victoria O. Shender, Ksenia G. Kuznetsova, Anna A. Kliuchnikova, and Sergei A. Moshkovskii. 2022. "Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy" International Journal of Molecular Sciences 23, no. 9: 5240. https://doi.org/10.3390/ijms23095240
APA StyleGoncharov, A. O., Shender, V. O., Kuznetsova, K. G., Kliuchnikova, A. A., & Moshkovskii, S. A. (2022). Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy. International Journal of Molecular Sciences, 23(9), 5240. https://doi.org/10.3390/ijms23095240