
Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy


Abstract
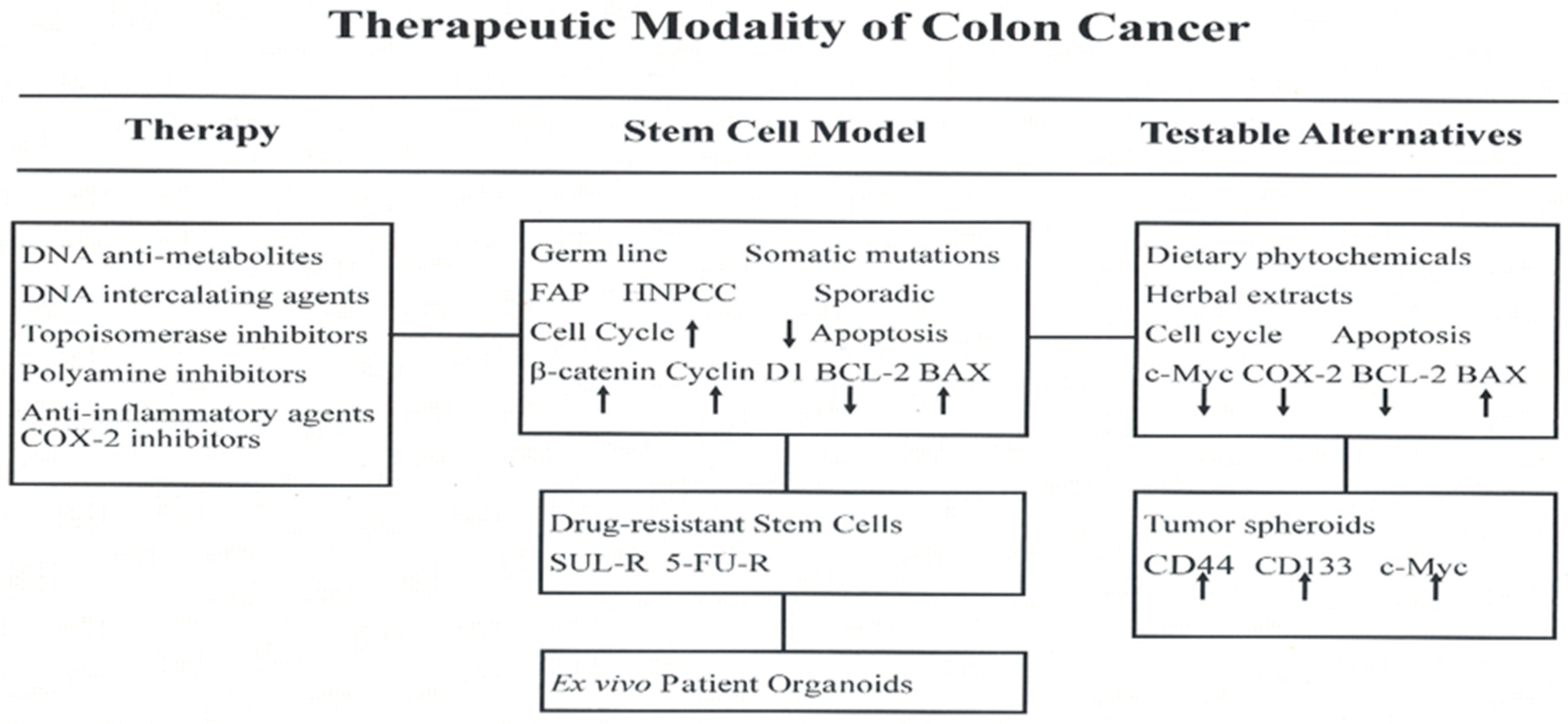
1. Introduction
2. Experimental Models
2.1. Mechanistic Assays
2.2. Test Agents
3. Carcinogenic Transformation
3.1. Growth Pattern and Molecular Markers in Colon Models
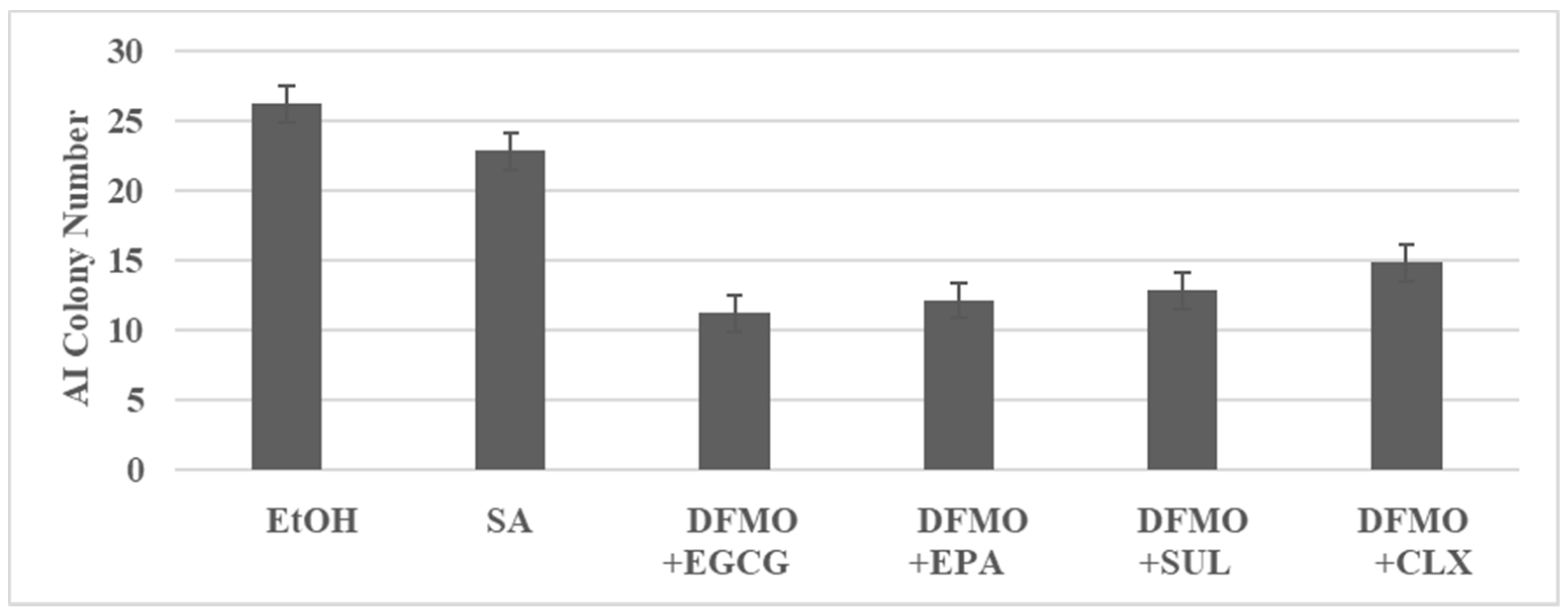
3.2. Combinatorial Anti-Proliferative Effects on FAP Model
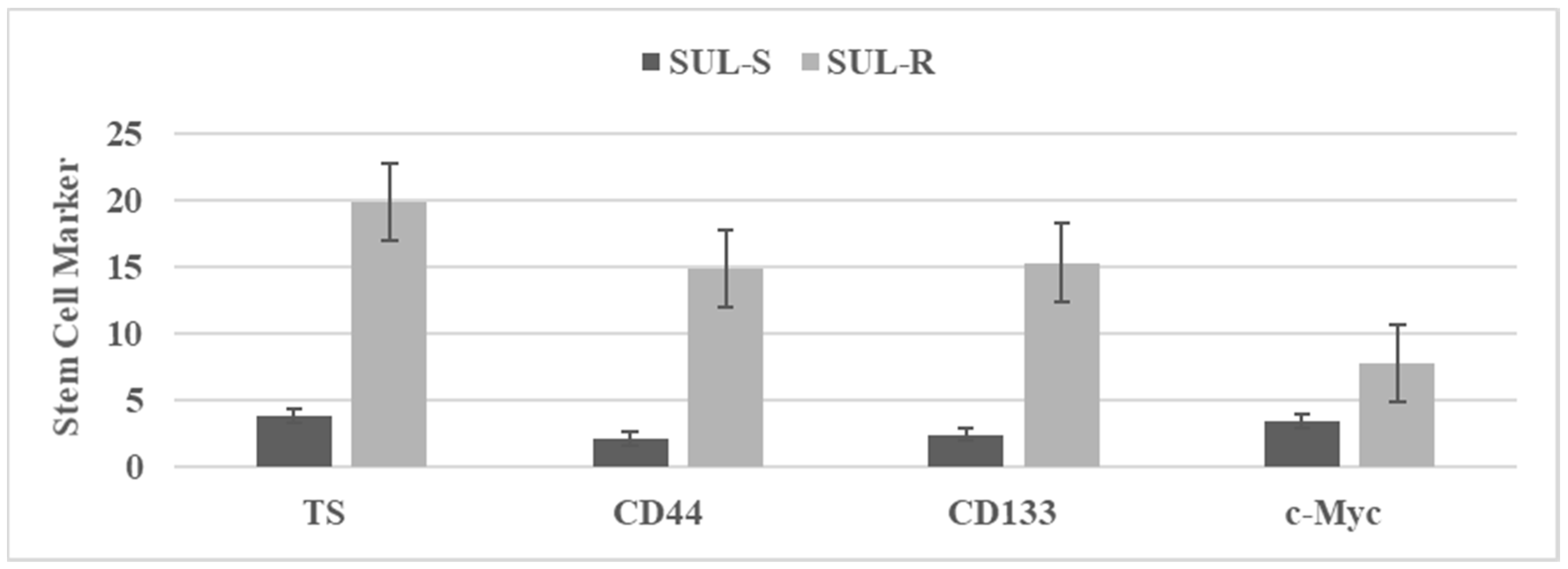
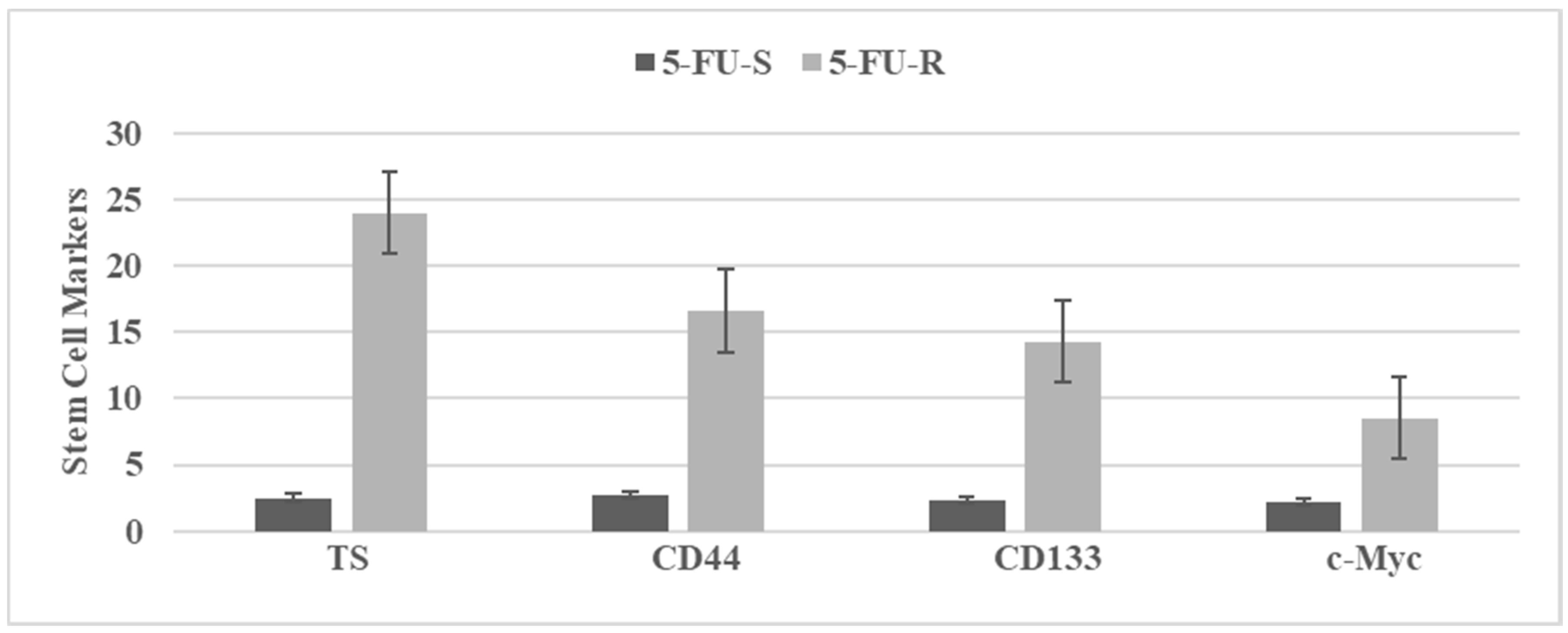
3.3. Characterization of Drug-Resistant Stem Cell Model
3.4. Tumor Spheroid Inhibition by Dietary Agents
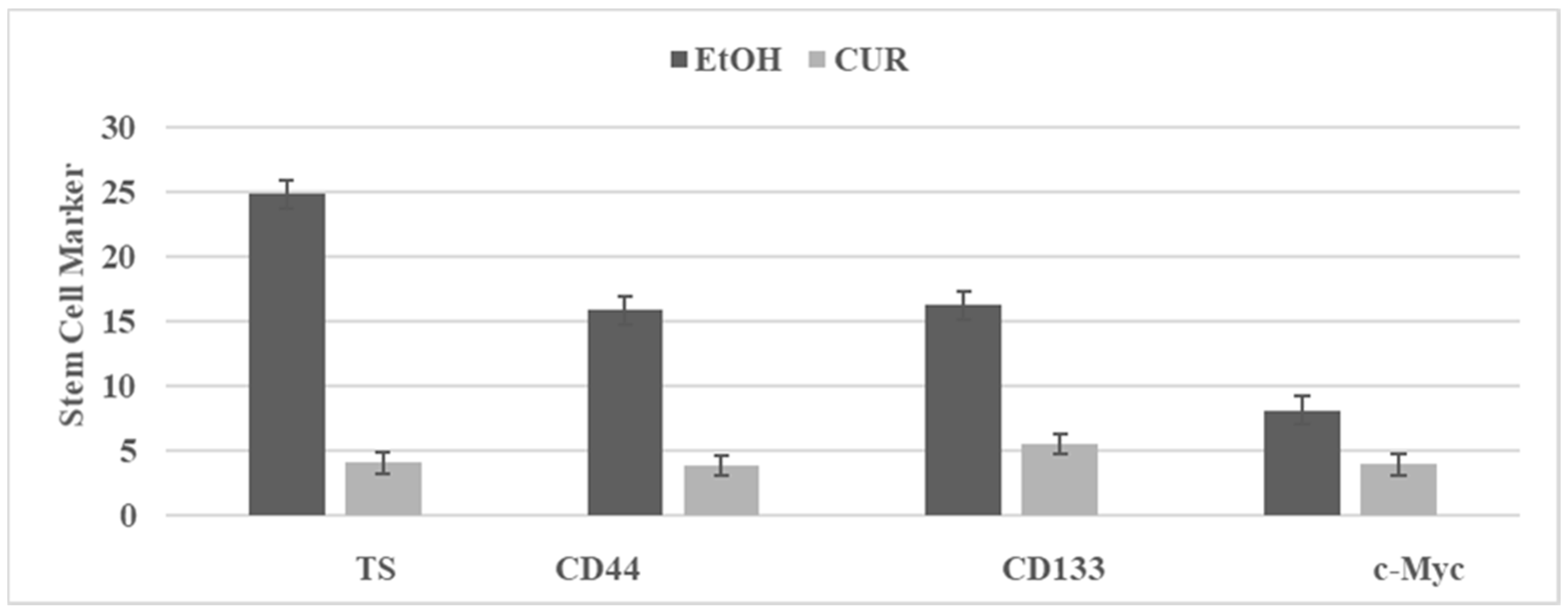
3.5. Effect of Curcumin on Stem Cells
4. Conclusions
5. Future Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society-Facts and Figures-2021; American Cancer Society: Atlanta, GA, USA, 2021.
- Williams, C.; Di Leo, A.; Niv, Y.; Gustafsson, J.Å. Estrogen receptor-β as a target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Filho, R.P.S.; Junior, S.A.; Begnami, M.D.; Ferreira, F.O.; Nakagawa, W.T. Estrogen receptor β as a prognostic marker of tumor progression in colorectal cancer with familial adenomatous polyps and sporadic polyps. Path. Oncol. Res. 2018, 24, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.L.; Javid, S.H.; Carothers, A.M.; Redston, M.; Bertagnolli, M.M. Estrogen receptor α and β are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Res. 2007, 67, 2366–2372. [Google Scholar] [CrossRef]
- Saleiro, D.; Murillo, G.; Lubahn, D.B.; Kopelovich, L.; Korach, K.S.; Mehta, R.G. Enhanced induction of mucin-depleted foci in estrogen receptor-β knockout mice. Cancer Prev. Res. (Phila.) 2010, 3, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Rustgi, A. The genetics of hereditary colon cancer. Genes Develop. 2007, 21, 2525–2538. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. 2021. Available online: http://www.nccn.org (accessed on 10 February 2022).
- Boolbol, S.K.; Dannenberg, A.J.; Chadburn, A.; Martucci, C.; Guo, X.-J.; Ramonetti, J.T.; Abreu-Goris, M.; Newmark, H.L.; Lipkin, M.L.; DeCosse, J.J.; et al. Cyclo-oxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996, 56, 2556–2560. [Google Scholar]
- Jacoby, R.F.; Siebert, K.; Cole, C.E.; Kellloff, G.; Lubet, R.A. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the Min mouse model of adenomatous polyposis. Cancer Res. 2000, 60, 5040–5044. [Google Scholar]
- Giardiello, F.M.; Hamilton, S.R.; Krush, A.J.; Piantadisis, S.; Hylind, L.M.; Celano, P.; Booker, S.V.; Robinson, C.R.; Offerhaus, G.J.A. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med. 1993, 328, 1313–1316. [Google Scholar] [CrossRef]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.; Verschoyle, R.D.; Hill, F.; Parveen, I.; Threadgil, M.D.; Sharma, R.A.; Williams, M.L.; Steward, W.P.; Gescher, A.J. Chemo-preventive efficacy and pharmacokinetics of Curcumin in the Min/+ mouse, a model for familial adenomatous polyposis. Cancer Epidemiol. Biomark. Prev. 2002, 11, 535–540. [Google Scholar]
- Ju, J.; Hong, J.; Zhou, J.-N.; Pan, Z.; Bose, M.; Liao, J.; Yang, G.; Liu, Y.Y.; Hou, Z.; Lin, Y.; et al. Inhibition of intestinal tumorigenesis in APC Min/+ mice by (−)-Epigallocatecin-3-gallate, the major catechin in green tea. Cancer Res. 2005, 65, 10623–10631. [Google Scholar] [CrossRef]
- Telang, N.; Li, G.; Katdare, M.; Sepkovic, D.W.; Bradlow, H.L.; Wong, G.Y.C. The nutritional herb Epimedium grandiflorum inhibits the growth in in a model for the Luminal A molecular subtype of breast cancer. Oncol. Lett. 2017, 13, 2477–2482. [Google Scholar] [CrossRef]
- Telang, N.; Nair, H.B.; Wong, G.Y.C. Growth inhibitory efficacy of Cornus officinalis in a cell culture model for triple-negative breast cancer. Oncol. Lett. 2019, 17, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Telang, N.; Nair, H.B.; Wong, G.Y.C. Growth inhibitory efficacy of the nutritional herb Psoralea corylifolia in a model for triple-negative breast cancer. Int. J. Func. Nutr. 2021, 2, 8. [Google Scholar] [CrossRef]
- Telang, N. Anti-inflammatory drug resistance selects putative cancer stem cells in a cellular model for genetically predisposed colon cancer. Oncol. Lett. 2018, 15, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Telang, N. Natural phytochemicals as testable therapeutic alternatives for HER-2-enriched breast cancer (Review). World Acad. Sci. J. 2020, 2, 19. [Google Scholar] [CrossRef]
- Telang, N. Stem cell models for genetically predisposed colon cancer (Review). Oncol. Lett. 2020, 20, 138. [Google Scholar] [CrossRef]
- Telang, N. Isolation and characterization of chemo-resistant stem cells from a mouse model of hereditary non-polyposis colon cancer. Stem Cells Cloning Adv. Appl. 2021, 14, 19–25. [Google Scholar] [CrossRef]
- Telang, N.; Katdare, M. Combinatorial prevention of carcinogenic risk in a model for familial colon cancer. Oncol. Rep. 2007, 17, 909–914. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Telang, N.; Katdare, M. Novel cell culture model for prevention of carcinogenic risk in familial adenomatous polyposis syndrome. Oncol. Rep. 2009, 21, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Edelmann, W.; Yang, K.; Van Leuwen, C.; Carlson, C.; Renault, B.; Breukel, C.; Alt, E.; Lipkin, M.; Khan, P.M. A targeted chain termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 8969–8973. [Google Scholar] [CrossRef] [PubMed]
- Katdare, M.; Kopelovich, L.; Telang, N. Efficacy of chemo-preventive agents on growth inhibition of Apc [+/−] 1638N COL colonic epithelial cells. Int. J. Mol. Med. 2002, 10, 427–432. [Google Scholar]
- Edelmann, W.; Yang, K.; Karaguchi, M.; Heyer, J.; Kneitz, M.L.B. Tumorigenesis in Mlh1 and Mlh1//1638N nice. Cancer Res. 1999, 59, 1301–1307. [Google Scholar]
- Gerner, E.W.; Meyskens, F.L. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 785–792. [Google Scholar] [CrossRef]
- Goldberg, R.M. Therapy of metastatic colon cancer. Oncologist 2006, 11, 981–987. [Google Scholar] [CrossRef][Green Version]
- Corpet, D.E.; Pierre, F. Point: From animal models to prevention of colon cancer. Systemic review of chemo-prevention in Min mice and choice of the model system. Cancer Epid. Biomark. Prev. 2003, 12, 391–400. [Google Scholar]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (-)-Epigallocatchin-3-Gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef]
- Jacoby, R.F.; Cole, C.E.; Tutsch, F.; Newton, M.A.; Kelloff, G.; Hawk, E.T.; Lubet, R.A. Chemo-preventive efficacy of combined piroxicam and difluoro methyl ornithine treatment on APC mutant Min mouse adenoma and selective toxicity against APC mutant embryos. Cancer Res. 2000, 60, 1864–1870. [Google Scholar] [PubMed]
- Swamy, M.V.; Cooma, I.; Patlolla, J.M.R.; Simi, B.; Reddy, B.S.; Rao, C.V. Modulation of cyclooxygenase-2 activities by the combined action of celecoxib and docosahexaenoic acid: Novel strategies for colon cancer prevention and treatment. Mol. Cancer Ther. 2004, 3, 215–221. [Google Scholar] [PubMed]
- Swamy, M.V.; Patlolla, J.M.R.; Steele, V.E.; Kopelovich, J.; Reddy, B.S.; Rao, C.V. Chemo-prevention of familial adenomatous polyposis by low doses of Atorvastatin and celecoxib. Given individually and in combination to APC Min/+ mice. Cancer Res. 2006, 66, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, Q.; Yu, L.; Zhu, J.; Cao, Y.; Gao, X. The signaling pathway and targets of traditional Chinese medicine and natural medicine in triple-negative breast cancer. J. Ethnopharmacol. 2021, 264, 113249. [Google Scholar] [CrossRef]
- Telang, N.; Nair, H.B.; Wong, G.Y.C. Anti-proliferative and pro-apoptotic effects of Dipsacus asperoides in a cellular model for triple-negative breast cancer. Arch. Breast Cancer 2022, 9, 66–75. [Google Scholar] [CrossRef]
- Aiello, P.; Sharghi, M.; Mansourkhani, S.M.; Ardekan, A.P.; Jouybari, L.; Daraei, N.; Peiro, K.; Mohamadian, S.; Rezaei, M.; Heidari, M.; et al. Medicinal plants in the prevention and treatment of colon cancer. Oxidat. Med. Cell. Longev. 2019, 2019, 2075614. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Kim, J.; Thi, H.T.H.; Bang, O.-S.; Lee, W.S.; Hong, S. Evaluation of anti-tumorigenic activity of BP3B against colon cancer with patient-derived tumor xenograft model. BMC Complement. Alt. Med. 2016, 16, 473. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.-Y.; Hong, S.; Shin, S.; Kim, Y.A.; Kim, N.S.; Bang, O.-S. A new herbal formula BP10A exerted an anti-tumor effect and enhanced anti-cancer effect of irinotecan and oxaliplatin in the colon cancer PDTX model. Biomed. Pharmacother. 2019, 116, 108987. [Google Scholar] [CrossRef]
- Li, Y.; Pu, R.; Zhou, L.; Wang, D.; Li, X. Effects of a chlorogenic acid-containing herbal medicine (LAS NB) on colon cancer. Evid. Based Complement. Altern. Med. 2021, 2021, 9923467. [Google Scholar] [CrossRef]
- Soteriou, D.; Fuchs, Y. A matter of life and death: Stem cell survival in tissue regeneration and tumor formation. Nat. Rev. Cancer 2018, 18, 187–201. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, L.; Qing, Q.; Li, Y.; Li, L.; Dong, X.; Xiao, B. Gene expression profile of cancer stem-like cells in the SW480 colon adenocarcinoma cell line. Oncol. Rep. 2019, 42, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Gheytanchi, E.; Naseri, M.; Karimi-Busheri, F.; Atyabi, F.; Mirsharif, E.S.; Bozorgmehr, M.; Ghods, R.; Madjd, Z. Morphological and molecular characteristics of spheroid formation in HT-29 and Caco-2 colorectal cancer cell lines. Cancer Cell Int. 2021, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Dalebra, P.; Dylla, S.J.; Park, I.K.; Luiu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollet, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumor growth in immune-deficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pillozi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Tan, H.Y.; Li, S.; Cheung, F.; Wang, N.; Nagamatsu, T.; Feng, Y. Cancer stem cells: The potential targets of Chinese medicines and their active compounds. Int. J. Mol. Sci. 2016, 17, 893. [Google Scholar] [CrossRef]
- Asharfizdeh, M.; Ahmadi, Z.; Mohamamdinejad, R.; Yaribeygi, H.; Serban, M.-C.; Orafai, H.M.; Sahebkar, A. Curcumim therapeutic modulation of the Wnt signaling pathway. Curr. Pharm. Biotechnol. 2020, 21, 1006–1015. [Google Scholar] [CrossRef]
- Manogaran, P.; Umapathy, D.; Karthikeyan, M.; Venkatachalam, K.; Singharavelu, A. Dietary phytochemicals as a potential source for targeting cancer stem cells. Cancer Investig. 2021, 39, 349–368. [Google Scholar] [CrossRef]
- Meerson, A.; Khatib, S.; Mahajna, J. Natural products targeting cancer stem cells for augmenting cancer therapeutics. Int. J. Mol. Sci. 2021, 22, 13044. [Google Scholar] [CrossRef]
- Naujokat, C.; Mc Kee, D.L. The “Big Five” phytochemicals targeting cancer stem cells: Curcumin, EGCG, sulforaphane, resveratrol and genistein. Curr. Med. Chem. 2021, 28, 4321–4342. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Lui, L.; Kalifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Shemmer, P.; Buchler, M.W.; Zoller, M.; et al. Synergistic activity of sorafenib and sulforphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 2010, 70, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Kalifatidis, G.; Baumann, B.; Rausch, V.; Mettern, J.; Gladkich, J.; Giese, N.; Moldenhauer, G.; Wirth, T.; Buchler, M.W.; et al. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int. J. Oncol. 2010, 37, 551–561. [Google Scholar] [CrossRef]
- Kalifatidis, G.; Labsch, S.; Rausch, V.; Mettern, J.; Gladkich, J.; Moldenhauer, G.; Buchler, M.W.; Salnikov, A.V.; Herr, I. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol. Ther. 2011, 19, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Soares, F.; Wang, S.; Wong, C.C.; Chen, H.; Yang, Z.; Liu, W.; Go, M.Y.Y.; Ahmed, M.; Zeng, Y.; et al. CRISPR screens identify cholesterol biosynthesis as a therapeutic target on stemness and drug resistance of colon cancer. Oncogene 2021, 40, 6601–6613. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient derived gastric carcinoma tumor growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef]
- Castro, N.P.; Rangel, M.C.; Merchant, A.S.; MacKnnon, G.; Cuttitta, F.; Salomon, D.S.; Kim, Y.S. Sulforaphane suppresses the growth of triple-negative breast cancer stem-like cells in vitro and in vivo. Cancer Prev. Res. (Phila.) 2019, 12, 147–158. [Google Scholar] [CrossRef]
- Kim, S.H.; Singh, S.V. Role of Kruppel-like factor-4 /p21CIP1 axis in breast cancer stem-like cell inhibition by benzyl isothiocyanate. Cancer Prev. Res. (Phila.) 2019, 12, 125–134. [Google Scholar] [CrossRef]
- Drost, J.; Van Jaarsveld, R.H.; Ponsoen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Crespo, M.; Villar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nature Med. 2017, 23, 878–884. [Google Scholar] [CrossRef]
- Drorst, J.; Celvers, H. organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.A.M.; Drew, D.A.; Dampier, C.H.; Plummer, S.J.; Eaton, S.; Bryant, J.; Díez-Obrero, V.; Mo, J.; Kedrin, D.; Zerjav, D.C.; et al. Transcriptome-wide in vitro effects of aspirin on patient-derived normal colon organoids. Cancer Prev. Res. (Phila.) 2021, 14, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.; Lanauze, C.; Wang, X.; Hayer, K.E.; Torres-Diz, M.; Leu, N.A.; Sela, Y.; Stanger, B.Z.; Lengner, C.J.; Thomas-Tikhonenko, A. Myc hyper-activates Wnt signaling in APC/CTNNB1-mutated colorectal cancer cells through miR-92a-dependent regression of DKK3. Mol. Cancer Res. 2021, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Bridges, A.E.; Ramachandran, S.; Tamizhmani, K.; Parwal, U.; Lester, A.; Rajpurohit, P.; Morera, D.S.; Hasanali, S.L.; Arjunan, P.; Jedeja, R.N.; et al. Rad51AP1 loss attenuates colorectal cancer stem cell renewal and sensitizes to chemotherapy. Mol. Cancer Res. 2021, 19, 1486–1497. [Google Scholar] [CrossRef]
- Bishnupuri, K.S.; Sainathan, S.K.; Ciorba, M.A.; Houchen, C.W.; Diechgraefe, B.K. Reg4 interacts with CD44 to regulate proliferation and stemness of colorectal and pancreatic cancer cells. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Altundag, O.; Canpinar, H.; Celebi-Saltic, B. Methionine affects the expression of pluripotency genes and protein levels associated with methionine metabolism in adult, fetal and cancer stem cells. J. Cell. Biochem. 2021, 123, 406–416. [Google Scholar] [CrossRef]
- Mehta, R.G. Functional significance of selective expression of ER-α and ER-β in mammary gland organ culture. Int. J. Mol. Sci. 2021, 22, 13151. [Google Scholar] [CrossRef]
- Fodde, R.; Smits, R.; Clevers, H. APC signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Ji, Y.; Lv, J.; Sun, D.; Huang, Y. Therapeutic strategies targeting Wnt/β-catenin signaling for colorectal cancer (Review). Int. J. Mol. Med. 2021, 49, 1. [Google Scholar] [CrossRef]
- Wester, R.A.; Voorthuijsen, L.V.; Neikes, H.K.; Dijkstra, J.J.; Lamers, L.A.; Frölich, S.; van der Sande, M.; Logie, C.; Lindeboom, R.G.; Vermeulen, M. Retinoic acid signaling drives differentiation toward the absorptive lineage in colorectal cancer. Science 2021, 24, 103444. [Google Scholar] [CrossRef]
- Ditonno, I.; Losurdo, G.; Rendina, M.; Pricci, M.; Girardi, B.; Lerardi, E.; Di Leo, A. Estrogen receptors in colorectal cancer: Facts, novelties and perspectives. Curr. Oncol. 2021, 28, 4256–4263. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Genotype | Origin a | Biological Characteristics | Model | Reference | |
|---|---|---|---|---|---|---|
| Mlh1 | Apc | |||||
| C57 COL | +/+ | +/+ | C57BL/6J | Immort. NT | Normal colon | [23] |
| 850 MIN COL | +/+ | +/− | Apc Min/+ | Immort. T | FAP | [23,24] |
| 1638N COL | +/+ | +/− | 1638N [+/−] | Immort. T | FAP | [27] |
| Mlh1 COL | +/− | +/+ | Mlh1 [+/−] | Immort. NT | HNPCC | [28] |
| Mlh1/1638N COL | +/− | +/− | Mlh1 [+/−] 1638N [+/−] | Immort. T | HNPCC | [22] |
| Agent | Source/Origin | References |
|---|---|---|
| Carnosic Acid (CA) | Rosemary terpenoid | [20] |
| Celecoxib (CLX) | Synthetic COX-2 inhibitor | [11] |
| Curcumin (CUR) | Curcuma longa root | [14] |
| Difluoromethyl ornithine (DFMO) | Synthetic ODC inhibitor | [29] |
| Docosa hexaenoic Acid (DHA) | Fish oil N-3 PUFA | [33] |
| Eicosa pentaenoic Acid (EPA) | Fish oil N-3 PUFA | [34] |
| Epigallo catechin gallate (EGCG) | Green tea polyphenol | [15,32] |
| Sulindac (SUL) | Synthetic COX-1/COX-2 inhibitor | [10] |
| Marker | Cellular Models | |||
|---|---|---|---|---|
| C57 COL | 850 MIN COL | p Value | Relative to C57 COL | |
| PDT (h.) a | 34.0 ± 3.8 | 14.0 ± 1.6 | 0.032 | −58.9% |
| Sat. Den. (×105) b | 7.7 ± 0.5 | 67.9 ± 7.5 | 0.001 | +7.8x |
| G1: S + G2/M c | 3.1 ± 0.3 | 0.6 ± 0.2 | 0.010 | −80.0% |
| Aneuploidy (%) c | Not detected | 81.7 ± 9.1 | ||
| Sub G0 (%) c | 4.3 ± 0.1 | 0.8 ± 0.3 | 0.010 | −81.4% |
| AI Colonies d | ||||
| Incidence | 0/18 | 18/18 | ||
| Number | ---- | 18.9 ± 2.5 | ||
| Tumor | ||||
| Incidence | 0/10 | 8/10 | ||
| Latency | 24 weeks | 3–5 weeks | ||
| Marker a | Cellular Model | |||
|---|---|---|---|---|
| C57 COL | 850 MIN COL | p Value | Relative to C57 COL | |
| Apc | 17.6 ± 1.7 | Not detected | ||
| β-catenin | 4.7 ± 0.3 | 9.0 ± 0.8 | 0.001 | +91.5% |
| Cyclin D1 | 3.0 ± 0.1 | 16.1 ± 1.5 | 0.001 | +4.4x |
| c-Myc | 2.1 ± 0.2 | 6.6 ± 0.4 | 0.010 | +2.1x |
| COX-2 | 3.9 ± 0.4 | 12.8 ± 1.8 | 0.010 | +2.3x |
| Dietary Agent | Tumor Spheroid Number a | p Value | Relative to Solvent Control |
|---|---|---|---|
| EtOH | 24.8 ± 2.2 | ||
| CUR | 4.0 ± 0.4 | 0.010 | −83.9% |
| EGCG | 6.0 ± 1.3 | 0.030 | −68.8% |
| EPA | 6.4 ± 1.4 | 0.030 | −66.7% |
| DHA | 6.8 ± 1.4 | 0.030 | −64.5% |
| CA | 8.6 ± 1.9 | 0.042 | −55.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telang, N. Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 2519. https://doi.org/10.3390/ijms23052519
Telang N. Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy. International Journal of Molecular Sciences. 2022; 23(5):2519. https://doi.org/10.3390/ijms23052519
Chicago/Turabian StyleTelang, Nitin. 2022. "Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy" International Journal of Molecular Sciences 23, no. 5: 2519. https://doi.org/10.3390/ijms23052519
APA StyleTelang, N. (2022). Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy. International Journal of Molecular Sciences, 23(5), 2519. https://doi.org/10.3390/ijms23052519