Abstract
HLJ1 (also called DNAJB4) is a member of the DNAJ/Hsp40 family and plays an important role in regulating protein folding and activity. However, there is little information about the role of HLJ1 in the regulation of physiological function. In this study, we investigated the role of HLJ1 in blood coagulation using wild-type C57BL/6 mice and HLJ1-null (HLJ1-/-) mice. Western blot analysis and immunohistochemistry were used to assess the expression and distribution of HLJ1 protein, respectively. The tail bleeding assay was applied to assess the bleeding time and blood loss. A coagulation test was used for measuring the activity of extrinsic, intrinsic and common coagulation pathways. Thromboelastography was used to measure the coagulation parameters in the progression of blood clot formation. The results showed that HLJ1 was detectable in plasma and bone marrow. The distribution of HLJ1 was co-localized with CD41, the marker of platelets and megakaryocytes. However, genetic deletion of HLJ1 did not alter blood loss and the activity of extrinsic and intrinsic coagulation pathways, as well as blood clot formation, compared to wild-type mice. Collectively, these findings suggest that, although HLJ1 appears in megakaryocytes and platelets, it may not play a role in the function of blood coagulation under normal physiological conditions.
1. Introduction
Blood coagulation is a very delicate process including a series of rigorously controlled cellular and molecular events that ultimately produces a stable blood clot [1]. Physiologically, coagulation begins almost immediately after the vascular endothelial injury. Exposing blood to the subendothelial space triggers two processes, namely changes in platelets and the exposure of subendothelial tissue factor to plasma factor VII, which ultimately leads to the formation of cross-linked fibrin [2,3]. Platelets immediately form a plug at the injury site, and then blood coagulation factors other than factor VII simultaneously respond in a cascade to form fibrin strands to strengthen the platelet plug and thereby prevent the loss of blood [2,3,4]. It has been documented that coagulation pathways can be divided into “intrinsic” and “extrinsic” routes initiated by factor XII (FXII) and FVIIa/tissue factor (TF), respectively. These two routes are integrated into the downstream “common” pathway at the FXa/FVa (prothrombinase) complex [5]. Pathologically, misfolded protein aggregates stimulate the activation of FXII but without inducing coagulation [6]. For example, the aggregated amyloid β peptide (Aβ) activates FXII in vitro, and the level of activated FXII is elevated in patients with Alzheimer’s disease (AD) and causes coagulation dysfunction [7]. Chaperones are a family of proteins that play a crucial role in the conformational folding or unfolding of proteins and assembly or disassembly with intracellular macromolecular structures [8,9]. Several lines of evidence demonstrate that chaperones are also important in regulating the activity of coagulation factors [10,11]. However, the molecular mechanism underlying the regulation of chaperones in blood coagulation is not fully understood.
HLJ1, an endogenous Src inhibitor, is also a member of the human heat shock protein (HSP) 40 subfamily and acts as a tumor suppressor in the pathogenesis of lung, colon, liver and gastric cancers [12,13,14]. HLJ1 inhibits cell invasion and impedes cell cycle progression by regulating the p21waf1 pathway in hepatocellular carcinoma and suppresses lung cancer progression by inhibiting Src activity [15,16]. Under physiological conditions, HSPs serve as molecular chaperones and regulate the stabilization of unfolded proteins [17]. In mammals, HSPs have been divided into six subfamilies according to their molecular size: HSP100, HSP90, HSP70, HSP60, HSP40 and small HSPs [18]. In addition to the relation to heat shock, HSPs are involved in the regulation of cellular functions upon stimulation with stimuli such as nutrient deprivation, oxidative stress and pathogen infection [19,20,21]. Recent studies have identified the role of HSPs in the blood coagulation of mammals [22,23]. For example, HSP72 promotes platelet aggregation, and HSP20 plays a vital role in coagulation function [24,25]. While the role of HLJ1 in cancer biology has been extensively investigated, its significance in blood coagulation is still elusive.
Given the physiological function of HSPs in protein folding and blood coagulation, we aimed to investigate the role of HLJ1 in blood coagulation in mice. We first investigated the expression of HLJ1 in bone marrow and plasma and then examined whether the genetic deletion of HLJ1 in mice affects blood coagulation. Our findings suggest that HLJ1 is expressed in megakaryocytes and platelets; however, genetic loss of HLJ1 function did not change the coagulation time, blood loss and the parameters of blood clot formation compared to wild-type (WT) mice.
2. Results
2.1. HLJ1 Is Expressed in the Bone Marrow and Plasma of Mice
To clarify the potential role of HLJ1 in regulating blood coagulation, we used WT and HLJ1-/- mice as our animal models. We first examined the effect of the genetic deletion of HLJ1 on body weight and tissue weight in mice, and our results showed that loss of HLJ1 function increased the body weight, heart weight and white adipose tissue (WAT) weight; however, there was no difference in the weights of liver, spleen, lung, kidney, brown adipose tissue (BAT), gastrocnemius muscle and brain (Table 1). Moreover, there was no difference in the peripheral blood counts of WT and HLJ1-/- mice (Table 2).

Table 1.
Body and tissue weights of WT and HLJ1-/- mice.
Table 2.
Peripheral blood counts of WT and HLJ1-/- mice.
We next examined the expression of HLJ1 in the plasma and bone marrow of mice. The results showed that HLJ1 protein appeared in the platelet-rich plasma of WT mice as a soluble form; however, it was undetectable in that of HLJ1-/- mice (Figure 1A). We then isolated platelets from plasma and found that no soluble form of HLJ1 was detected in platelet-poor plasma; in contrast, the protein expression of HLJ1 in plasma was mainly restricted in platelets, as evidenced by the expression of HLJ1 being co-localized with CD41, the marker for platelets and megakaryocytes (Figure 1B,C,E). In addition, we found that the protein expression of HLJ1 in bone marrow mainly appeared in megakaryocytes and platelets, as evidenced by the expression of HLJ1 being co-localized with CD41 in the bone marrow of WT mice (Figure 1D,F).
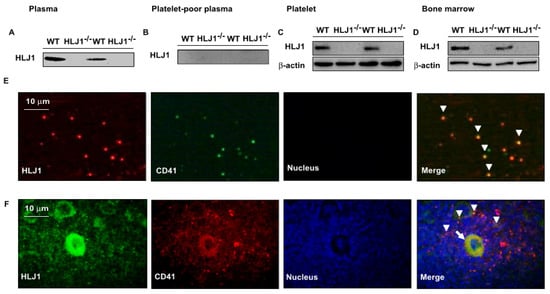
Figure 1.
Expression of HLJ in plasma and bone marrow of WT and HLJ1-/- mice. (A–D) Western blot analysis of HLJ1 in plasma, platelet-poor plasma, platelets and bone marrow. (E) Representative images of HLJ1 expression in platelets (arrowheads) of plasma by immunohistochemistry staining. (F) Representative images of HLJ1 expression in platelets (arrowheads) and megakaryocytes (arrow) of bone marrow by immunohistochemistry staining.
2.2. The Role of HLJ1 in the Bleeding Time and Blood Loss in Mice
To evaluate the effect of HLJ1 on bleeding time and blood loss, we used HLJ1-/- mice as our animal model. The results showed that the genetic disruption of HLJ1 did not affect the bleeding time (Figure 2A,B) and blood loss (Figure 2C,D) compared to WT mice.
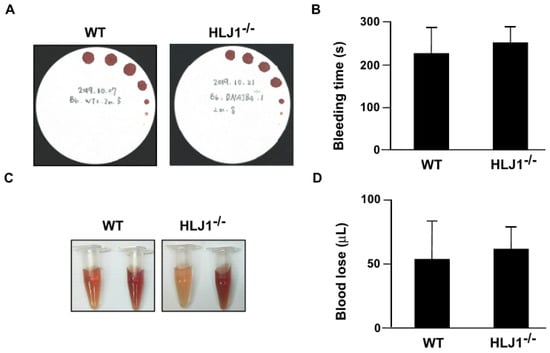
Figure 2.
Tail bleeding time and blood loss in WT and HLJ1-/- mice. (A,B) Tail bleeding time was assessed by the filter paper method in WT and HLJ1-/- mice. (C,D) Blood loss after tail transection was measured in WT and HLJ1-/- mice. Data are the mean ± SEM from 9 mice.
2.3. The Role of HLJ1 in the Activity of Blood Coagulation in Mice
To investigate whether HLJ1 plays a role in regulating the blood coagulation response, we measured the coagulation parameters in WT mice and HLJ1-/- mice. The results showed that the genetic deletion of HLJ1 in mice had no effect on the activity of intrinsic and extrinsic time (PT and aPTT, Figure 3A,B), the content of fibrinogen (FIB) and the time to fibrin clot formation (fibrinogen activity test) as compared to WT mice (Figure 3C,D).
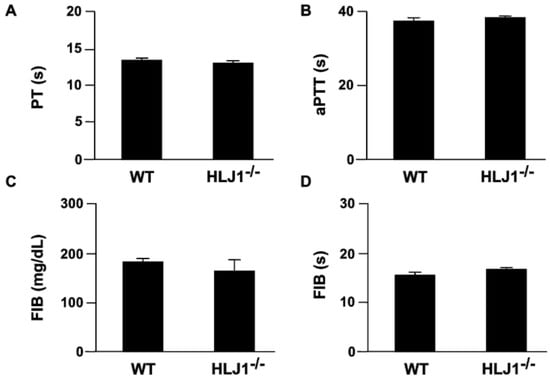
Figure 3.
The parameters of plasma coagulation in WT and HLJ1-/- mice. (A,B) The plasma of mice was isolated, and prothrombin time (PT) and activated partial thromboplastin time (aPTT) were evaluated. (C,D) The plasma level of fibrinogen (FIB) and time of fibrin clot formation (s) were assessed. Data are the mean ± SEM from 9 mice.
We further applied thromboelastography (TEG) to examine the role of HLJ1 in the dynamic real-time reaction of coagulation. The results demonstrated that there was no difference in the TEG parameters, including R, the time to formation of initial fibrin threads; K, the time until the clot reaches a certain strength; α angle, the rapidity with which the clot forms; maximum amplitude (MA), the clot’s maximum strength; amplitude (A), the end time amplitude; and the coagulation index, the overall clotting activity of HLJ1-/- mice compared to WT mice (Figure 4A–H).
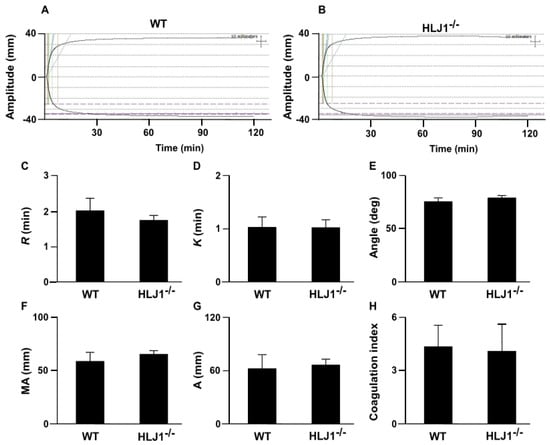
Figure 4.
Thromboelastography (TEG) tracing in WT and HLJ1-/- mice. (A,B) Representative tracing depicting the rate of clot formation and degradation was performed in WT and HLJ1-/- mice. (C–H) The reaction time (R), kinetics time (K), α angle, maximum amplitude (MA), the end time amplitude and the coagulation index (A) were analyzed. Data are the mean ± SEM from 9 mice.
3. Discussion
HLJ1, a chaperone protein, plays a crucial role in regulating protein folding stability and quality control [12,13,14]; however, the biological significance of HLJ1 in blood coagulation has not been fully investigated yet. In this study, we used WT and HLJ1-/- mice as our in vivo model and characterized the potential effect of HLJ1 on blood coagulation. Our results suggest that HLJ1 indeed appeared in the megakaryocytes and platelets of the bone marrow and plasma of WT mice. Platelets are known to play an important role in regulating hemostasis, inflammation and wound healing and are thus implicated in the pathogenesis of human diseases under certain conditions [26,27]. However, the genetic disruption of HLJ1 did not change the platelet count of peripheral blood, the activity of blood coagulation and the formation of blood clots as compared to WT mice. Based on these observations, we concluded that HLJ1, a member of the HSP40 subfamily, is not involved in the activation of the coagulation system. Nevertheless, further investigations regarding the role of HLJ1 in the biological functions of platelets other than hemostasis are warranted. Whether HLJ1 plays an important role under specific stress induction should be further investigated.
Increasing evidence suggests that HSPs are expressed in platelets and participate in the regulation of platelet function and coagulation function [28,29]. For instance, Li et al. reported that HSP20 plays a vital role in coagulation function and decreased HSP20 might contribute to the pathogenesis of pre-eclampsia [25]. Zhu et al. reported that phosphorylation of HSP27 by thrombin is crucial for cytoskeletal rearrangements in the process of platelet activation [28]. HSP70 regulates platelet integrin activation, granule secretion and aggregation [29]. Suzuki et al. demonstrated that HSP72 promotes activator-induced platelet aggregation [24]. Very recently, Jackson et al. (2020) reported that the inhibition of HSP40 and HSP90 by pharmacological antagonists abolishes the dense granule release, thromboxane synthesis and aggregation in thrombin-treated platelets [30]. The evidence from these studies indicates that HSPs play an important role in platelet function and coagulation function; however, their regulatory mechanism is not fully understood. To this end, further investigation to delineate the molecular mechanisms of HSPs in platelet activation and blood coagulation is warranted.
To our knowledge, information about the role of HLJ1 in human diseases is limited. HLJ1 reportedly serves as a tumor suppressor and inhibits the proliferation, migration and invasion of cancer cells by regulating the STAT1/p21 pathway and could thus be a biomarker for cancers [15]. However, its role in other human diseases is largely unknown. In this study, we found that genetic deletion of HLJ1 caused a significant increase in body weight, which could be attributed to the increase in heart weight and WAT weight, suggesting that HLJ1 may play a key role in the regulation of heart diseases and metabolic diseases. Several lines of evidence indicate that the level of HSPs is correlated with the pathogenesis of cardiac hypertrophy [31,32,33,34]. Kumarapeli et al. have reported that the small HSP αB-crystallin (CryAB) is critical for normal cardiac function, and overexpression of CryAB suppresses pressure-overload-induced cardiac hypertrophy and delays the development of cardiac failure [31,32]. Nevertheless, whether HLJ1 is involved in the pathogenesis of cardiovascular diseases and its molecular regulation are unclear. To this end, further investigations for clarifying the role of HLJ1 in cardiovascular diseases and the underlying mechanisms are warranted.
Importantly, HSPs are involved in the pathogenesis of lipid disorders, including obesity, non-alcoholic fatty liver disease (NAFLD), insulin resistance and atherosclerosis [33,34,35,36,37,38,39,40,41,42]. Di Naso et al. reported that HSP70-mediated anti-inflammation is impaired in obese patients, which contributes to the development of NAFLD [35]. Chung et al. demonstrated that HSP72 protects against obesity-induced insulin resistance by blocking inflammation [36]. Circulating HSP70 acts as an inflammatory mediator and accelerates the progression of atherosclerosis [37]. Moreover, Gungor et al. reported that nuclear HSP70 has a cholesterol-lowering effect by activating the liver X receptor pathway [38]. The findings from these studies suggest that HSPs play an important role in the regulation of lipid metabolism and related metabolic diseases. This notion is further supported by our findings that genetic deletion of HLJ1 causes an increase in WAT weight. However, further investigations for delineating whether HLJ1 is involved in metabolic diseases and thermogenesis and its underlying molecular mechanism are required.
Nevertheless, our study contains several limitations. HSPs are a large family of evolutionarily conserved molecular chaperones that regulate protein folding and maintain the protein structure and function in response to pathophysiological and environmental stimuli [18,43,44]. In this study, however, we did not use disease models to mimic the pathology of diseases and delineate the potential role and mechanism of HLJ1 in blood coagulation. Moreover, we do not have clinical data to support our observations from in vivo studies. Therefore, further pathological studies or clinical trials describing the implications of HLJ1 in coagulation disorders induced by various diseases are warranted.
In conclusion, this study clarifies the relationship between HLJ1 and blood coagulation. Although HLJ1 is expressed in platelets and megakaryocytes, it has no role in the regulation of blood coagulation under physiological conditions; in contrast, HLJ1 may play a certain role in the regulation of cardiac function and lipid metabolism. Here, information should be provided for a better understanding of the function of HLJ1 in platelet biology and blood coagulation.
4. Materials and Methods
4.1. Reagents
Rabbit antibodies for β actin and HLJ1 (13064-1-AP) were obtained from Proteintech (Rosemont, IL, USA). Mouse antibody for CD41 (553847) was obtained from BD Bioscience (San Jose, CA, USA). NH4OH solution was obtained from PanReac AppliChem (Darmstadt, Germany). Prothrombin time (PT) activated partial thromboplastin time (aPTT) and fibrinogen (FIB) reagents were purchased from TECO Medical Instruments (Neufahrn, NB, Germany).
4.2. Animal
This study conformed to the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, Eighth Edition, 2011), and all animal experiments were approved by the Animal Care and Utilization Committee of the College of Medicine, National Taiwan University (20210257). HLJ1-/- C57BL/6 mice were generated using the gene targeting strategy. Specifically, exon2 of mouse HLJ1 was deleted by traditional homologous recombination in embryonic stem cells. HLJ1-/- mice were generated, and the polymerase chain reaction (PCR) of genomic DNA was performed to confirm the HLJ1-/- genotype. PCR was performed with the following primers: HLJ1 (forward): 5′-GGC TTG CTG TCT AAG GTG ATG-3′, HLJ1 (reverse): 5′-ACG TTC AAA GCT ATC ATA GTC-3′. Primers were used at 95 °C for 5 min, followed by 40 cycles at 95 °C for 30 s, 62 °C for 30 s, 72 °C 30 s and additional extension at 72 °C for 5 min. Male wild-type (WT) C57BL/6 mice and HLJ1-/- mice from the Laboratory Animal Center of the National Taiwan University College of Medicine (Taipei, Taiwan) were housed in barrier facilities on a 12-h light/12-h dark cycle. Five mice were group-housed per cage and fed with a regular chow diet containing 4.5% fat by weight and 0.02% cholesterol (Newco Distributors, Redwood, CA, USA). Two-month-old WT and HLJ1-/- mice were euthanized with CO2 after measuring the body weight of mice. Heart, liver, spleen, lung, kidney, muscle, WAT and BAT were isolated and weighed. Plasma was isolated from citrated blood (3.2% sodium citrate, 1:10) by centrifugation for 10 min at 1500 g and 4 °C and then stored at −80 °C until further analysis.
4.3. Immunohistochemistry
Femur and tibia were isolated and fixed with formalin. After decalcification, the samples were embedded in paraffin. The blocks were then cut into 8 μm sections and subjected to histological examination. The deparaffinized sections were blocked with 2% BSA for 30 min at 37 °C. Then, sections were incubated with the primary antibody and with the corresponding secondary antibody overnight at 4 °C. Photomicrographs were digitally captured under a Leica DM IRB fluorescence microscope (Leica Microsystems, Philadelphia, PA, USA).
4.4. Western Blot Analysis
Bone marrow cells from mice were lysed, and the total protein was extracted. Aliquots of protein (50 μg) or plasma (2 μL) were separated on 12% SDS gels and then transblotted onto a PVDF membrane (Millipore, Bedford, MA). After being blocked with 5% skim milk, the blotting membrane was incubated with the primary antibodies, followed by the corresponding horseradish peroxidase-conjugated secondary antibodies. Bands were visualized using an enzyme-linked chemiluminescence detection kit (PerkinElmer, Waltham, MA, USA), and the band density was measured using quantitative software (TotalLab, Newcastle upon Tyne, UK).
4.5. Tail Bleeding Assay
Mice were anesthetized using pentobarbital (50 mg/kg) and then positioned horizontally on a platform to let the tail hang down naturally. A distal 3-mm segment of the tail was cut off with a scalpel. To evaluate bleeding from the incision, filter paper (Kotobuki, Tokyo, Japan) was used to touch the edge of the forming clot every 30 s over a period of 600 s. The dried filter paper was scanned to measure the blood loss. To dissolve the hemoglobin, the filter papers were dipped into 0.04% NH4OH solution for 2 h. The level of hemoglobin was assessed by a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) at 416 nm. The volume of blood in each sample was calculated as compared to the standard curve, which was obtained by dropping defined volumes of WT mouse blood onto a filter paper and extracting hemoglobin as described above.
4.6. Coagulation Test
The activation of extrinsic and intrinsic coagulation pathways was examined using PT, aPTT and FIB assay kits according to the manufacturer’s instructions. The activity of PT, aPTT and FIB was assessed using an analyzer (TECO Medical Instruments, Neufahrn NB, Germany).
4.7. Thromboelastography (TEG)
Mice were killed by CO2, and blood samples were isolated by cardiac puncture. For TEG analysis, 340 μL citrated blood was mixed with 20 μL CaCl2 (0.2 M) and then subjected to the Heamoscope TEG 5000 Thrombelastograph Hemostasis Analyzer (Haemonetics Corp., Braintree, MA, USA) according to the manufacturer’s instructions. The major coagulation parameters of TEG were assessed, including R, the time to the formation of initial fibrin threads; K, the time until the clot reaches a certain strength; α angle, the rapidity with which the clot forms; maximum amplitude, the clot’s maximum strength; amplitude, the end time amplitude; and coagulation index, the overall clotting activity.
4.8. Statistical Analysis
The results are presented as the mean ± SEM. The Mann–Whitney U test was used to compare two independent groups. Differences were considered statistically significant at p < 0.05. The Shapiro–Wilk test was used for testing normality. The p-value for each group was at p > 0.05, and there was no significant difference. SPSS software v8.0 (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses.
Author Contributions
Conceptualization, K.-Y.S. and T.-S.L.; methodology, M.-C.H., W.-J.L., B.-C.G., P.-A.H., Y.-H.T. and C.-H.C.; formal analysis, M.-C.H.; investigation, M.-C.H., W.-J.L., B.-C.G. and C.-H.C.; data curation, M.-C.H. and B.-C.G.; writing—original draft preparation, M.-C.H. and T.-S.L.; supervision, T.-S.L.; funding acquisition, T.-S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (106-2320-B-002-057-MY3, 106-2320-B-002-056, 106-2811-B-002-146; 108-2811-B-002-542, 108-2320-B-002-032-MY3 and 110-2314-B-002-269). We thank the staff of the imaging core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Balestra, D.; Branchini, A. Molecular mechanisms and determinants of innovative correction approaches in coagulation factor deficiencies. Int. J. Mol. Sci. 2019, 20, 3036. [Google Scholar] [CrossRef] [PubMed]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism action of platelets and crucial blood coagulation pathways in hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 8, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Mertens, K.; Bertina, R.M. Activation of human coagulation factor VIII by activated factor X, the common product of the intrinsic and the extrinsic pathway of blood coagulation. Thromb. Haemost. 1982, 47, 96–100. [Google Scholar] [CrossRef]
- Maas, C.; Govers-Riemslag, J.W.; Bouma, B.; Schiks, B.; Hazenberg, B.P.; Lokhorst, H.M.; Hammarström, P.; ten Cate, H.; de Groot, P.G.; Bouma, B.N.; et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J. Clin. Investig. 2008, 118, 3208–3218. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Renné, T.; Strickland, S. The Alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Foit, L.; George, J.S.; Zhang, B.W.; Brooks, C.L.; Bardwell, J.C. Chaperone activation by unfolding. Proc. Natl. Acad. Sci. USA 2013, 110, E1254–E1262. [Google Scholar] [CrossRef]
- Poothong, J.; Pottekat, A.; Siirin, M.; Campos, A.R.; Paton, A.W.; Paton, J.C.; Lagunas-Acosta, J.; Chen, Z.; Swift, M.; Volkmann, N.; et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood 2020, 135, 1899–1911. [Google Scholar] [CrossRef]
- Pignani, S.; Todaro, A.; Ferrarese, M.; Marchi, S.; Lombardi, S.; Balestra, D.; Pinton, P.; Bernardi, F.; Pinotti, M.; Branchini, A. The chaperone-like sodium phenylbutyrate improves factor IX intracellular trafficking and activity impaired by the frequent p.R294Q mutation. J. Thromb. Haemost. 2018, 16, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, J.; Zhang, C.; Fu, W.; Xiao, X.; Ruan, S.; Zhang, Y.; Luo, X.; Tang, M. HLJ1 is a novel biomarker for colorectal carcinoma progression and overall patient survival. Int. J. Clin. Exp. Pathol. 2014, 7, 969–977. [Google Scholar] [PubMed]
- Zhang, L.; Cai, X.; Chen, K.; Wang, Z.; Wang, L.; Ren, M.; Huang, A.; Tang, H. Hepatitis B virus protein up-regulated HLJ1 expression via the transcription factor YY1 in human hepatocarcinoma cells. Virus Res. 2011, 157, 76. [Google Scholar] [CrossRef] [PubMed]
- Simões-Correia, J.; Silva, D.I.; Melo, S.; Figueiredo, J.; Caldeira, J.; Pinto, M.T.; Girão, H.; Pereira, P.; Seruca, R. DNAJB4 molecular chaperone distinguishes WT from mutant E-cadherin, determining their fate in vitro and in vivo. Hum. Mol. Genet. 2014, 23, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Wang, C.C.; Chang, G.C.; Chen, C.Y.; Chen, H.Y.; Cheng, C.L.; Yang, Y.P.; Wu, C.Y.; Shih, F.Y.; Liu, C.C.; et al. A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J. Natl. Cancer Inst. 2006, 98, 825–838. [Google Scholar] [CrossRef]
- Chen, C.H.; Chang, W.H.; Su, K.Y.; Ku, W.H.; Chang, G.C.; Hong, Q.S.; Hsiao, Y.J.; Chen, H.C.; Chen, H.Y.; Wu, R.; et al. HLJ1 is an endogenous Src inhibitor suppressing cancer progression through dual mechanisms. Oncogene 2016, 35, 5674–5685. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef]
- Khalil, A.A.; Kabapy, N.F.; Deraz, S.F.; Smith, C. Heat shock proteins in oncology: Diagnostic biomarkers or therapeutic targets? Biochim. Biophys. Acta 2011, 1816, 89–104. [Google Scholar] [CrossRef]
- Moura, C.S.; Lollo, P.C.B.; Morato, P.N.; Amaya-Farfan, J. Dietary nutrients and bioactive substances modulate heat shock protein (HSP) expression: A review. Nutrients 2018, 10, 683. [Google Scholar] [CrossRef]
- Feidantsis, K.; Giantsis, I.A.; Vratsistas, A.; Makri, S.; Pappa, A.Z.; Drosopoulou, E.; Anestis, A.; Mavridou, E.; Exadactylos, A.; Vafidis, D.; et al. Correlation between intermediary metabolism, Hsp gene expression, and oxidative stress-related proteins in long-term thermal-stressed Mytilus galloprovincialis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R264–R281. [Google Scholar] [CrossRef]
- Balasubramaniam, B.; Vinitha, T.; Deepika, S.; JebaMercy, G.; VenkataKrishna, L.M.; Balamurugan, K. Analysis of Caenorhabditis elegans phosphoproteome reveals the involvement of a molecular chaperone, HSP-90 protein during Salmonella enterica Serovar Typhi infection. Int. J. Biol. Macromol. 2019, 37, 620–646. [Google Scholar] [CrossRef] [PubMed]
- Polanowska-Grabowska, R.; Gear, A.R. Heat-shock proteins and platelet function. Platelets 2000, 11, 6–22. [Google Scholar] [CrossRef] [PubMed]
- İn, E.; Deveci, F.; Kaman, D. Assessment of heat shock proteins and endothelial dysfunction in acute pulmonary embolism. Blood Coagul. Fibrinolysis 2016, 27, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kosuge, Y.; Kobayashi, K.; Kurosaki, Y.; Ishii, N.; Aoyama, N.; Ishihara, K.; Ichikawa, T. Heat-shock protein 72 promotes platelet aggregation induced by various platelet activators in rats. Biomed. Res. 2017, 38, 175–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, F.; He, M.; Yang, M.; Fan, Y.; Chen, Y.; Xia, X.; Xie, Y.; Deng, D. Alteration of heat shock protein 20 expression in preeclamptic patients and its effect in vascular and coagulation function. Front. Med. 2018, 12, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019, 122, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Zhu, Y.; O’Neill, S.; Saklatvala, J.; Tassi, L.; Mendelsohn, M.E. Phosphorylated HSP27 associates with the activation-dependent cytoskeleton in human platelets. Blood 1994, 84, 3715–3723. [Google Scholar] [CrossRef]
- Rigg, R.A.; Healy, L.D.; Nowak, M.S.; Mallet, J.; Thierheimer, M.L.; Pang, J.; McCarty, O.J.; Aslan, J.E. Heat shock protein 70 regulates platelet integrin activation, granule secretion and aggregation. Am. J. Physiol. Cell Physiol. 2016, 310, C568–C575. [Google Scholar] [CrossRef]
- Jackson, J.W.; Rivera-Marquez, G.M.; Beebe, K.; Tran, A.D.; Trepel, J.B.; Gestwicki, J.E.; Blagg, B.S.J.; Ohkubo, S.; Neckers, L.M. Pharmacologic dissection of the overlapping impact of heat shock protein family members on platelet function. J. Thromb. Haemost. 2020, 18, 1197–1209. [Google Scholar] [CrossRef]
- Kumarapeli, A.R.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Kumarapeli, A.R.; Horak, K.; Wang, X. Protein quality control in protection against systolic overload cardiomyopathy: The longterm role of small heat shock proteins. Am. J. Transl. Res. 2010, 2, 390–401. [Google Scholar] [PubMed]
- Xu, Q.; Li, D.G.; Holbrook, N.J.; Udelsman, R. Acute hypertension induces heat-shock protein 70 gene expression in rat aorta. Circulation 1995, 92, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Patton, W.F.; Erdjument-Bromage, H.; Marks, A.R.; Tempst, P.; Taubman, M.B. Components of the protein synthesis and folding machinery are induced in vascular smooth muscle cells by hypertrophic and hyperplastic agents. Identification by comparative protein phenotyping and microsequencing. J. Biol. Chem. 1995, 270, 21404–21410. [Google Scholar] [CrossRef] [PubMed]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.; de Bittencourt, P.I., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, E.V.; Chipigina, N.S.; Shurdumova, M.H.; Kovalenko, E.I.; Sapozhnikov, A.M. Heat shock protein 70 kDa as a target for diagnostics and therapy of cardiovascular and cerebrovascular diseases. Curr. Pharm. Des. 2019, 25, 710–714. [Google Scholar] [CrossRef]
- Gungor, B.; Vanharanta, L.; Hölttä-Vuori, M.; Pirhonen, J.; Petersen, N.H.T.; Gramolelli, S.; Ojala, P.M.; Kirkegaard, T.; Ikonen, E. HSP70 induces liver X receptor pathway activation and cholesterol reduction in vitro and in vivo. Mol. Metab. 2019, 28, 135–143. [Google Scholar] [CrossRef]
- Grundtman, C.; Kreutmayer, S.B.; Almanzar, G.; Wick, M.C.; Wick, G. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 960–968. [Google Scholar] [CrossRef]
- Habich, C.; Sell, H. Heat shock proteins in obesity: Links to cardiovascular disease. Horm. Mol. Biol. Clin. Investig. 2015, 21, 117–124. [Google Scholar] [CrossRef]
- Archer, A.E.; Von Schulze, A.T.; Geiger, P.C. Exercise, heat shock proteins and insulin resistance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160529. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B.; Lanaspa, M.A.; Johnson, R.J. The role of autoimmune reactivity induced by heat shock protein 70 in the pathogenesis of essential hypertension. Br. J. Pharmacol. 2019, 176, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting heat shock proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).