
5′RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa

 , , , and
, , , and 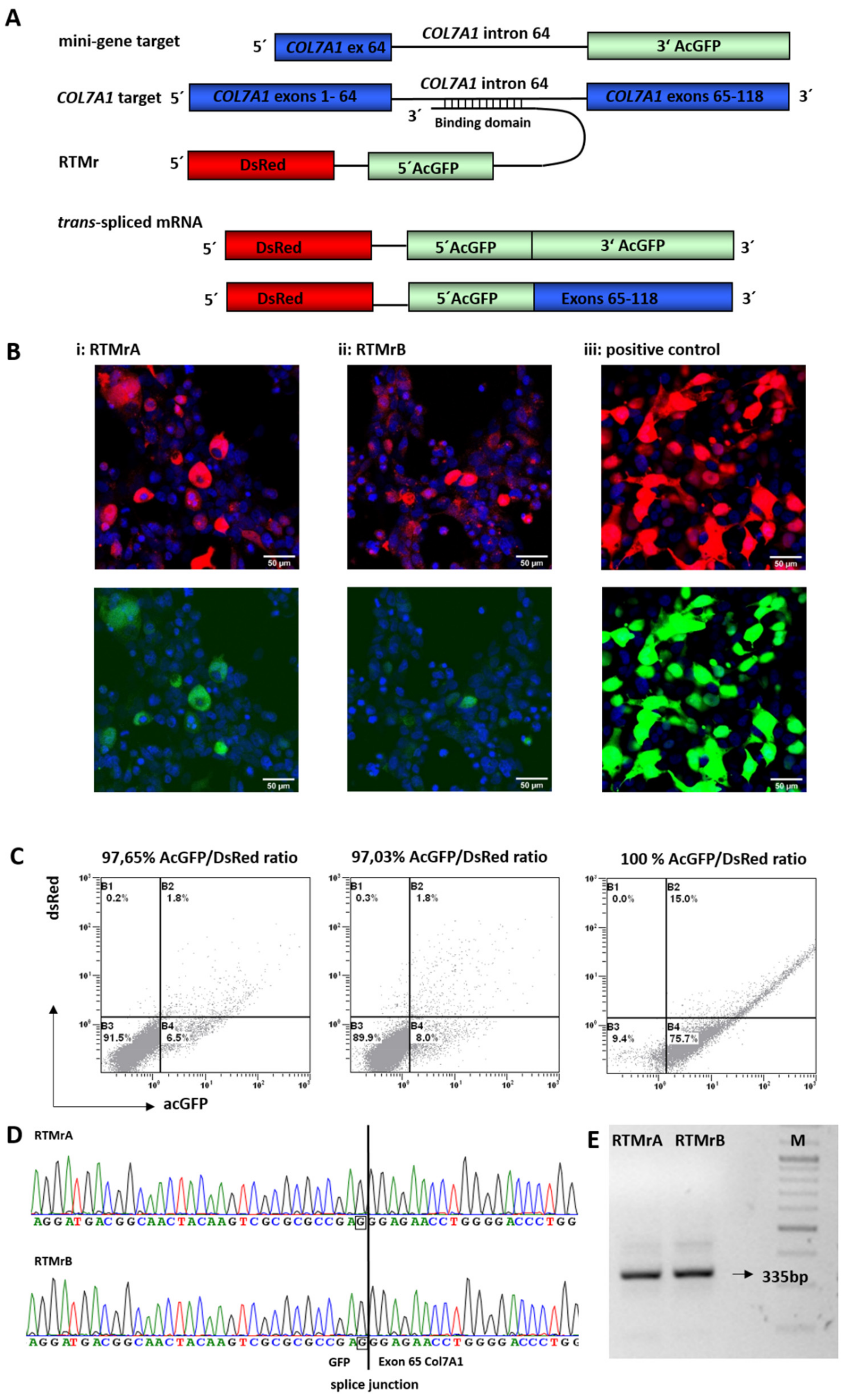{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Selection of Highly Functional RTMs Using a Fluorescence-Based Screening System
2.2. Endogenous 5′ Trans-Splicing in Keratinocytes
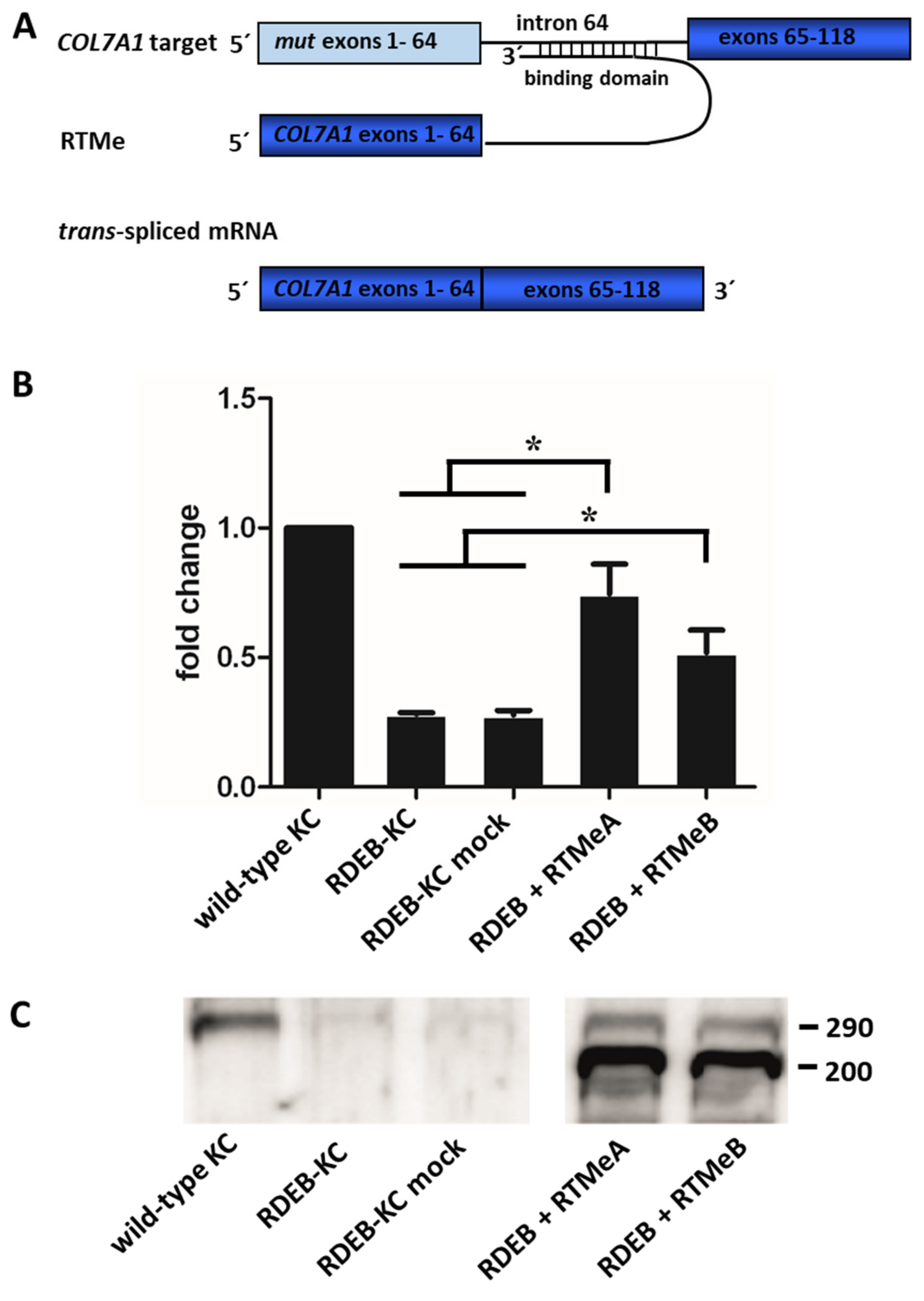
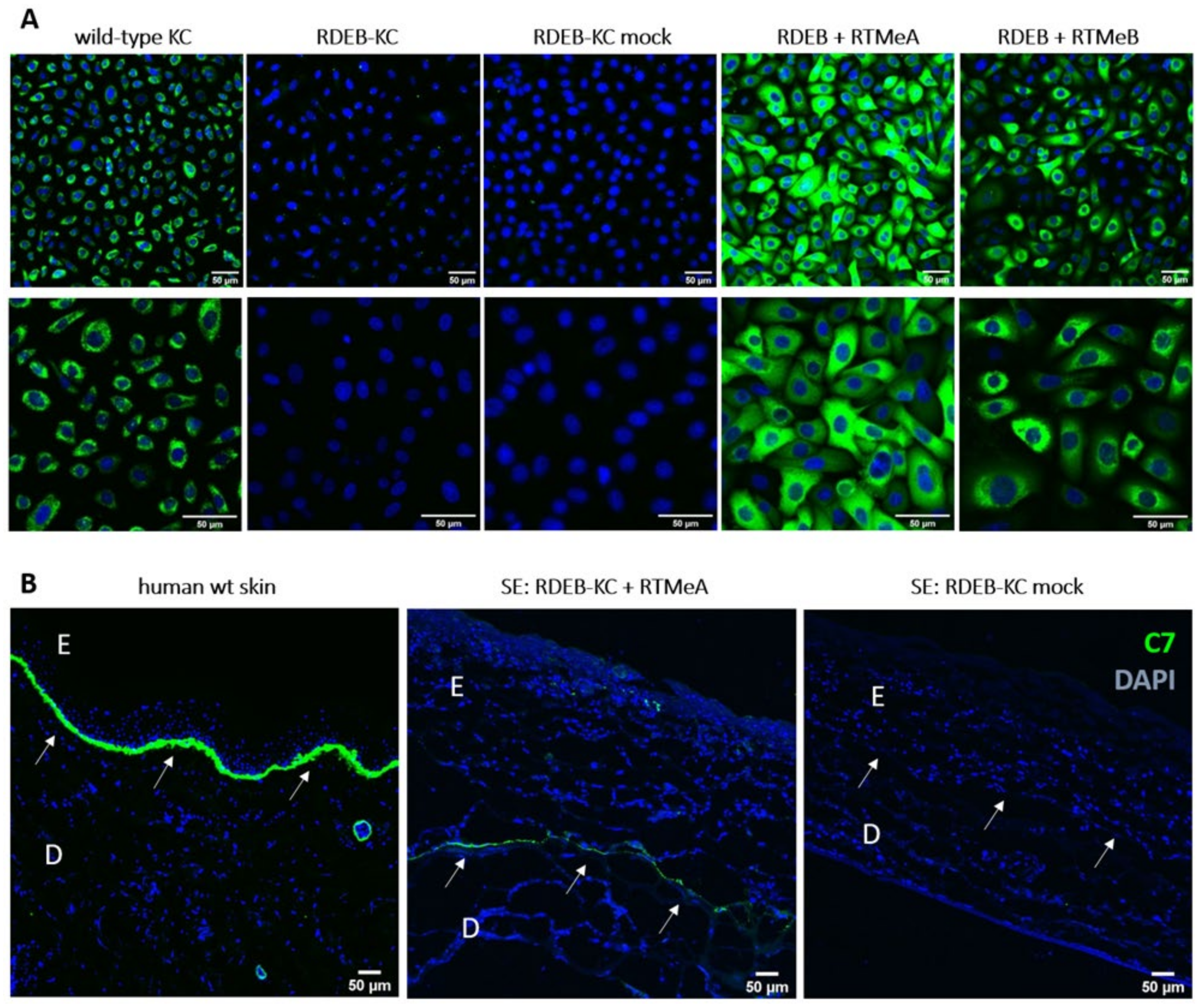
2.3. Endogenous Correction of COL7A1 in RDEB Patient Cells
2.4. C7 Expression in Skin Equivalents
3. Discussion
4. Materials and Methods
4.1. Cloning of Screening Constructs
4.2. Plasmid Transfection and Trans-Splicing Evaluation
4.3. RNA Isolation
4.4. cDNA Amplification and PCR
4.5. Cloning of Constructs for Endogenous Experiments
4.6. Viral Transduction
4.7. Semi-Quantitative (sq)RT-PCR
4.8. Western Blot Analysis
4.9. Immunofluorescence Staining of Type VII Collagen in Keratinocytes
4.10. Generation of Skin Equivalents
4.11. Immunofluorescence Staining of Type VII Collagen and Laminin 332 in Skin Equivalents
4.12. Cells
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Christiano, A.M.; Hoffman, G.G.; Chung-Honet, L.C.; Lee, S.; Cheng, W.; Uitto, J.; Greenspan, D.S. Structural Organization of the Human Type VII Collagen Gene (COL7A1), Composed of More Exons than Any Previously Characterized Gene. Genomics 1994, 21, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Uitto, J. Type VII Collagen: The Anchoring Fibril Protein at Fault in Dystrophic Epidermolysis Bullosa. Dermatol. Clin. 2010, 28, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. Journal of the American Academy of Dermatology 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Gache, Y.; Baldeschi, C.; Del Rio, M.; Gagnoux-Palacios, L.; Larcher, F.; Lacour, J.-P.; Meneguzzi, G. Construction of Skin Equivalents for Gene Therapy of Recessive Dystrophic Epidermolysis Bullosa. Hum. Gene Ther. 2004, 15, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Titeux, M.; Pendaries, V.; Zanta-Boussif, M.A.; Décha, A.; Pironon, N.; Tonasso, L.; Mejia, J.E.; Brice, A.; Danos, O.; Hovnanian, A. SIN Retroviral Vectors Expressing COL7A1 Under Human Promoters for Ex Vivo Gene Therapy of Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2010, 18, 1509–1518. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Nguyen, N.T.; Bezchinsky, M.Y.; Marinkovich, M.P.; Lane, A.T.; Khavari, P.A. Long-Term Type VII Collagen Restoration to Human Epidermolysis Bullosa Skin Tissue. Hum. Gene Ther. 2010, 21, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.S.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4, e130554. [Google Scholar] [CrossRef]
- Lwin, S.M.; Syed, F.; Di, W.-L.; Kadiyirire, T.; Liu, L.; Guy, A.; Petrova, A.; Abdul-Wahab, A.; Reid, F.; Phillips, R.; et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chen, M.; O’Toole, E.A.; Muellenhoff, M.; Medina, E.; Kasahara, N.; Woodley, D.T. Development and characterization of a recombinant truncated type VII collagen “minigene”. Implication for gene therapy of dystrophic epidermolysis bullosa. J. Biol. Chem. 2000, 275, 24429–24435. [Google Scholar] [CrossRef] [PubMed]
- Woodley, D.T.; Keene, D.R.; Atha, T.; Huang, Y.; Ram, R.; Kasahara, N.; Chen, M. Intradermal Injection of Lentiviral Vectors Corrects Regenerated Human Dystrophic Epidermolysis Bullosa Skin Tissue in Vivo. Mol. Ther. 2004, 10, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Sat, E.; Leung, K.H.; Bruckner-Tuderman, L.; Cheah, K.S.E. Tissue-specific expression and long-term deposition of human collagen VII in the skin of transgenic mice: Implications for gene therapy. Gene Ther. 2000, 7, 1631–1639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mecklenbeck, S.; Compton, S.H.; Mejia, J.E.; Cervini, R.; Hovnanian, A.; Bruckner-Tuderman, L.; Barrandon, Y. A mi-croinjected COL7A1-PAC vector restores synthesis of intact procollagen VII in a dystrophic epidermolysis bullosa keratinocyte cell line. Hum. Gene Ther. 2002, 13, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Murauer, E.M.; Gache, Y.; Gratz, I.K.; Klausegger, A.; Muss, W.; Gruber, C.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2011, 131, 74–83. [Google Scholar] [CrossRef]
- Tockner, B.; Kocher, T.; Hainzl, S.; Reichelt, J.; Bauer, J.W.; Koller, U.; Murauer, E.M. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther. 2016, 23, 775–784. [Google Scholar] [CrossRef]
- Peking, P.; Koller, U.; Duarte, B.; Murillas, R.; Wolf, S.; Maetzig, T.; Rothe, M.; Kocher, T.; García, M.; Brachtl, G.; et al. An RNA-targeted therapy for dystrophic epidermolysis bullosa. Nucleic Acids Res. 2017, 45, 10259–10269. [Google Scholar] [CrossRef]
- Horiuchi, T.; Aigaki, T. Alternative trans-splicing: A novel mode of pre-mRNA processing. Biol Cell 2006, 98, 135–140. [Google Scholar] [CrossRef]
- Koller, U.; Wally, V.; Bauer, J.W.; Murauer, E.M. Considerations for a Successful RNA Trans-splicing Repair of Genetic Disorders. Mol. Ther. Nucleic Acids 2014, 3, e157. [Google Scholar] [CrossRef]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-Mediated Trans-Splicing: The Therapeutic Cut and Paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef]
- Liemberger, B.; Hofbauer, J.P.; Wally, V.; Arzt, C.; Hainzl, S.; Kocher, T.; Murauer, E.M.; Bauer, J.W.; Reichelt, J.; Koller, U. RNA Trans-Splicing Modulation via Antisense Molecule Interference. Int. J. Mol. Sci. 2018, 19, 762. [Google Scholar] [CrossRef]
- Wally, V.; Brunner, M.; Lettner, T.; Wagner, M.; Koller, U.; Trost, A.; Murauer, E.M.; Hainzl, S.; Hintner, H.; Bauer, J.W. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum. Mol. Genet. 2010, 19, 4715–4725. [Google Scholar] [CrossRef]
- Peking, P.; Breitenbach, J.S.; Ablinger, M.; Muss, W.H.; Poetschke, F.J.; Kocher, T.; Koller, U.; Hainzl, S.; Kitzmueller, S.; Bauer, J.W.; et al. An ex vivo RNA trans-splicing strategy to correct human generalized severe epidermolysis bullosa simplex. Br. J. Dermatol. 2019, 180, 141–148. [Google Scholar] [CrossRef]
- Kierlin-Duncan, M.N.; Sullenger, B.A. Using 5′-PTMs to repair mutant beta-globin transcripts. RNA 2007, 13, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Klausegger, A.; Koller, U.; Lochmuller, H.; Krause, S.; Wiche, G.; Mitchell, L.G.; Hintner, H.; Bauer, J.W. 5′ trans-splicing repair of the PLEC1 gene. J. Invest. Dermatol. 2008, 128, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Zayed, H.; Xia, L.; Yerich, A.; Yant, S.R.; Kay, M.A.; Puttaraju, M.; McGarrity, G.J.; Wiest, D.L.; McIvor, R.S.; Tolar, J.; et al. Correction of DNA protein kinase deficiency by spliceosome-mediated RNA trans-splicing and sleeping beauty transposon delivery. Mol. Ther. 2007, 15, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mansfield, S.G.; Cote, C.A.; Du Jiang, P.; Weng, K.; Amar, M.J.; Brewer, B.H.; Remaley, A.T.; McGarrity, G.J.; Garcia-Blanco, M.A.; et al. Trans-splicing Into Highly Abundant Albumin Transcripts for Production of Therapeutic Proteins In Vivo. Mol. Ther. 2009, 17, 343–351. [Google Scholar] [CrossRef]
- Liu, X.; Luo, M.; Zhang, L.N.; Yan, Z.; Zak, R.; Ding, W.; Mansfield, S.G.; Mitchell, L.G.; Engelhardt, J.F. Spliceo-some-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum. Gene Ther. 2005, 16, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Mansfield, S.G.; Bartel, R.C.; Hiriyanna, S.; Mitchell, L.G.; Garcia-Blanco, M.A.; Walsh, C.E. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat. Med. 2003, 9, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Kathirvel, P.; Yee, W.C.; Lai, P.S. Correction of dystrophia myotonica type 1 pre-mRNA transcripts by artificial trans-splicing. Gene Ther. 2009, 16, 211–217. [Google Scholar] [CrossRef]
- Coady, T.H.; Lorson, C.L. Trans-Splicing-Mediated Improvement in a Severe Mouse Model of Spinal Muscular Atrophy. J. Neurosci. 2010, 30, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Dallinger, G.; Puttaraju, M.; Mitchell, L.G.; Yancey, K.B.; Yee, C.; Klausegger, A.; Hintner, H.; Bauer, J.W. Development of spliceosome-mediated RNA trans-splicing (SMaRT™) for the correction of inherited skin diseases. Exp. Dermatol. 2003, 12, 37–46. [Google Scholar] [CrossRef]
- Murauer, E.M.; Koller, U.; Hainzl, S.; Wally, V.; Bauer, J.W. A Reporter-Based Screen to Identify Potent 3′ Trans-Splicing Molecules for Endogenous RNA Repair. Hum. Gene Ther. Methods 2013, 24, 19–27. [Google Scholar] [CrossRef]
- Bauer, J.W.; Murauer, E.M.; Wally, V.; Koller, U. RNA Trans-Splicing for Genodermatoses. Mol. Dermatol. 2012, 961, 441–455. [Google Scholar] [CrossRef]
- Gruber, C.; Gratz, I.; Murauer, E.; Mayr, E.; Koller, U.; Bruckner-Tuderman, L.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Spliceosome-Mediated RNA Trans-Splicing Facilitates Targeted Delivery of Suicide Genes to Cancer Cells. Mol. Cancer Ther. 2011, 10, 233–241. [Google Scholar] [CrossRef]
- Gruber, C.; Koller, U.; Murauer, E.; Hainzl, S.; Hüttner, C.; Kocher, T.; South, A.P.; Hintner, H.; Bauer, J.W. The design and optimization of RNA trans-splicing molecules for skin cancer therapy. Mol. Oncol. 2013, 7, 1056–1068. [Google Scholar] [CrossRef]
- Goto, M.; Sawamura, D.; Nishie, W.; Sakai, K.; McMillan, J.R.; Akiyama, M.; Shimizu, H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dys-trophic epidermolysis bullosa patients. J. Investig. Dermatol. 2006, 126, 2614–2620. [Google Scholar] [CrossRef]
- Bornert, O.; Hogervorst, M.; Nauroy, P.; Bischof, J.; Swildens, J.; Athanasiou, I.; Tufa, S.F.; Keene, D.R.; Kiritsi, D.; Hainzl, S.; et al. QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Investig. Dermatol. 2020, 141, 883–893.e6. [Google Scholar] [CrossRef] [PubMed]
- Kühl, T.; Mezger, M.; Hausser, I.; Guey, L.T.; Handgretinger, R.; Bruckner-Tuderman, L.; Nyström, A. Collagen VII Half-Life at the Dermal-Epidermal Junction Zone: Implications for Mechanisms and Therapy of Genodermatoses. J. Investig. Dermatol. 2016, 136, 1116–1123. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayr, E.; Ablinger, M.; Lettner, T.; Murauer, E.M.; Guttmann-Gruber, C.; Piñón Hofbauer, J.; Hainzl, S.; Kaiser, M.; Klausegger, A.; Bauer, J.W.; et al. 5′RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa. Int. J. Mol. Sci. 2022, 23, 1732. https://doi.org/10.3390/ijms23031732
Mayr E, Ablinger M, Lettner T, Murauer EM, Guttmann-Gruber C, Piñón Hofbauer J, Hainzl S, Kaiser M, Klausegger A, Bauer JW, et al. 5′RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa. International Journal of Molecular Sciences. 2022; 23(3):1732. https://doi.org/10.3390/ijms23031732
Chicago/Turabian StyleMayr, Elisabeth, Michael Ablinger, Thomas Lettner, Eva M. Murauer, Christina Guttmann-Gruber, Josefina Piñón Hofbauer, Stefan Hainzl, Manfred Kaiser, Alfred Klausegger, Johann W. Bauer, and et al. 2022. "5′RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa" International Journal of Molecular Sciences 23, no. 3: 1732. https://doi.org/10.3390/ijms23031732
APA StyleMayr, E., Ablinger, M., Lettner, T., Murauer, E. M., Guttmann-Gruber, C., Piñón Hofbauer, J., Hainzl, S., Kaiser, M., Klausegger, A., Bauer, J. W., Koller, U., & Wally, V. (2022). 5′RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa. International Journal of Molecular Sciences, 23(3), 1732. https://doi.org/10.3390/ijms23031732