
Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance

Abstract
1. Introduction
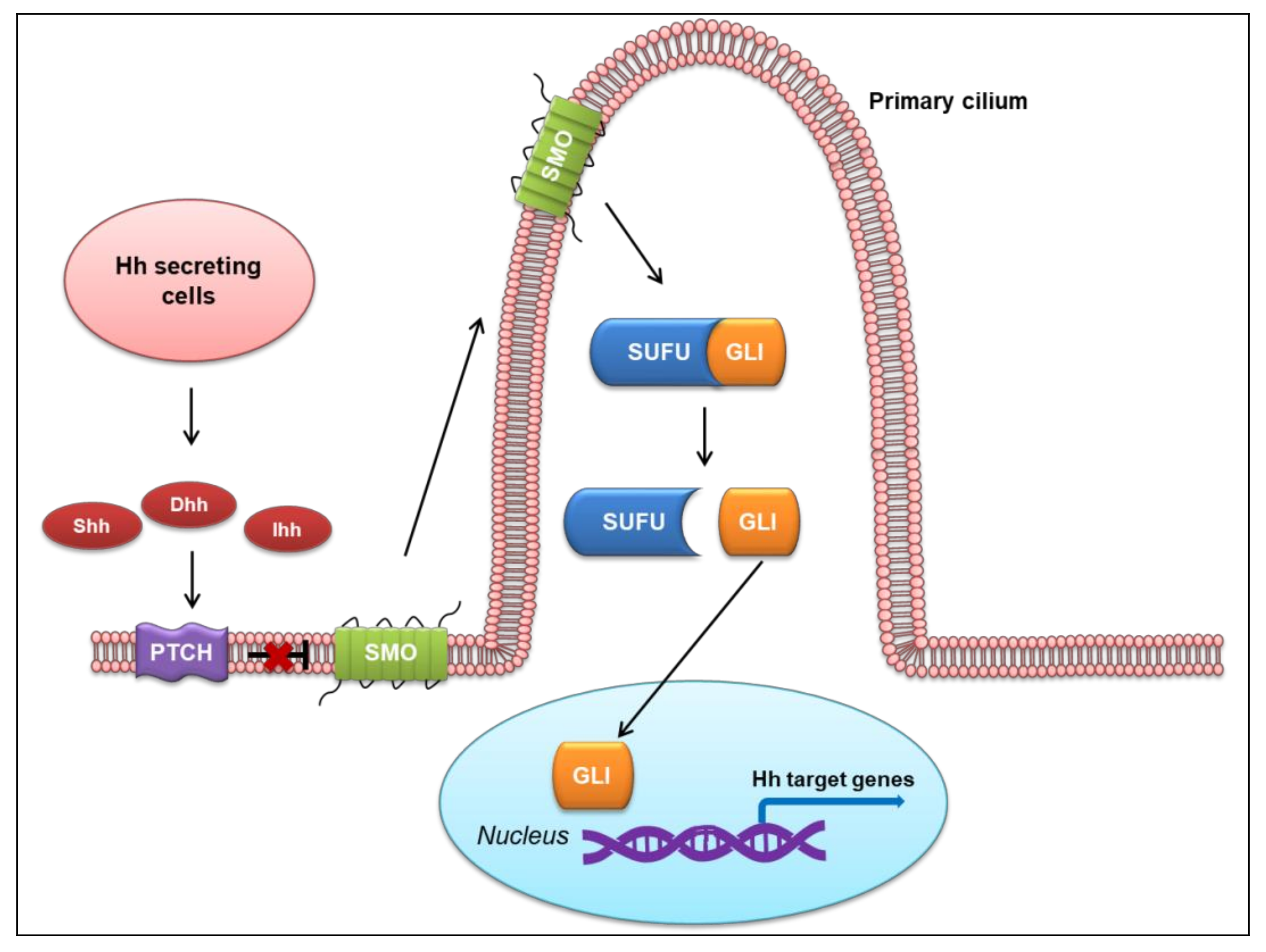
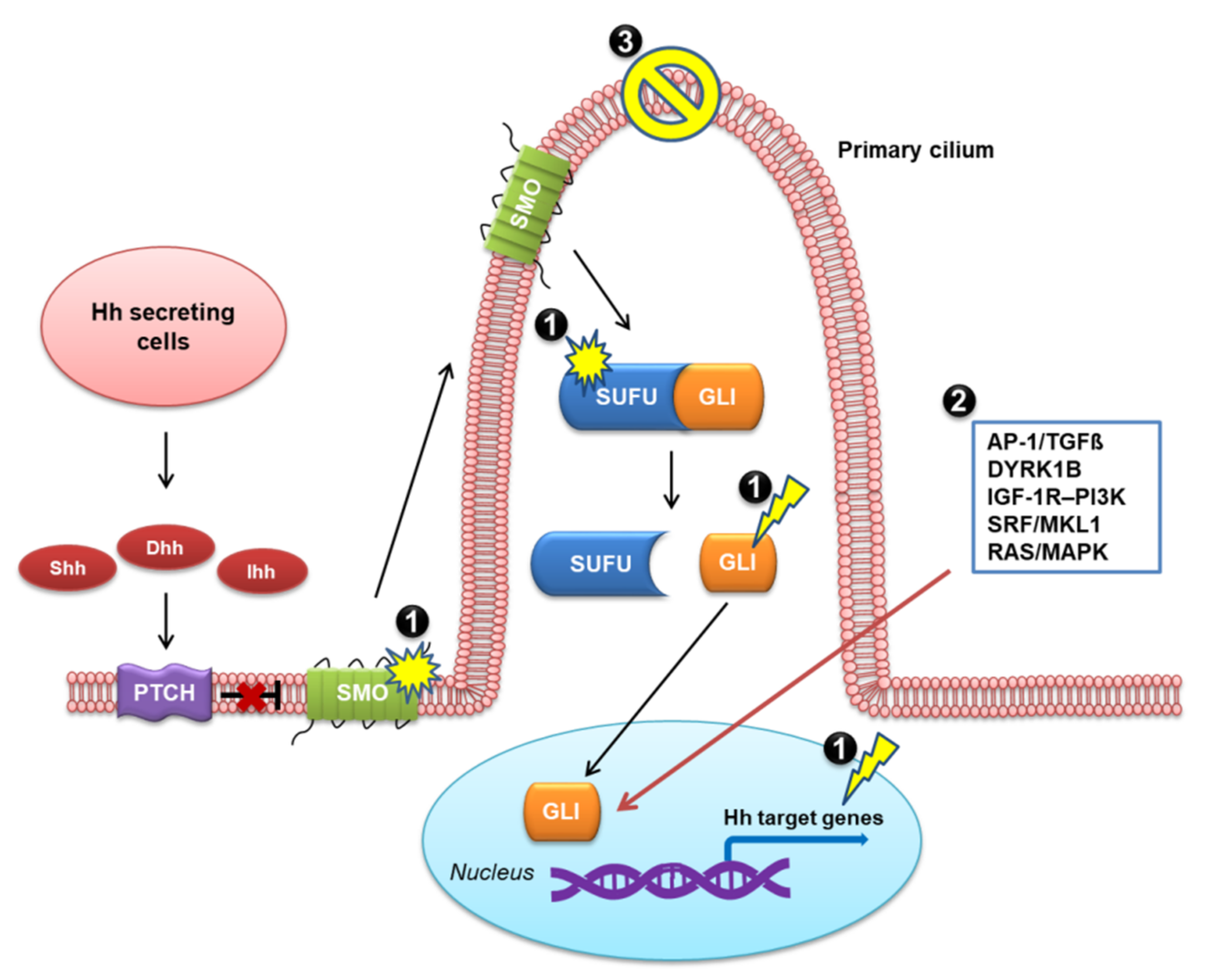
2. Overview of the Hh Signaling Pathway
2.1. Canonical Hh Signaling Pathway
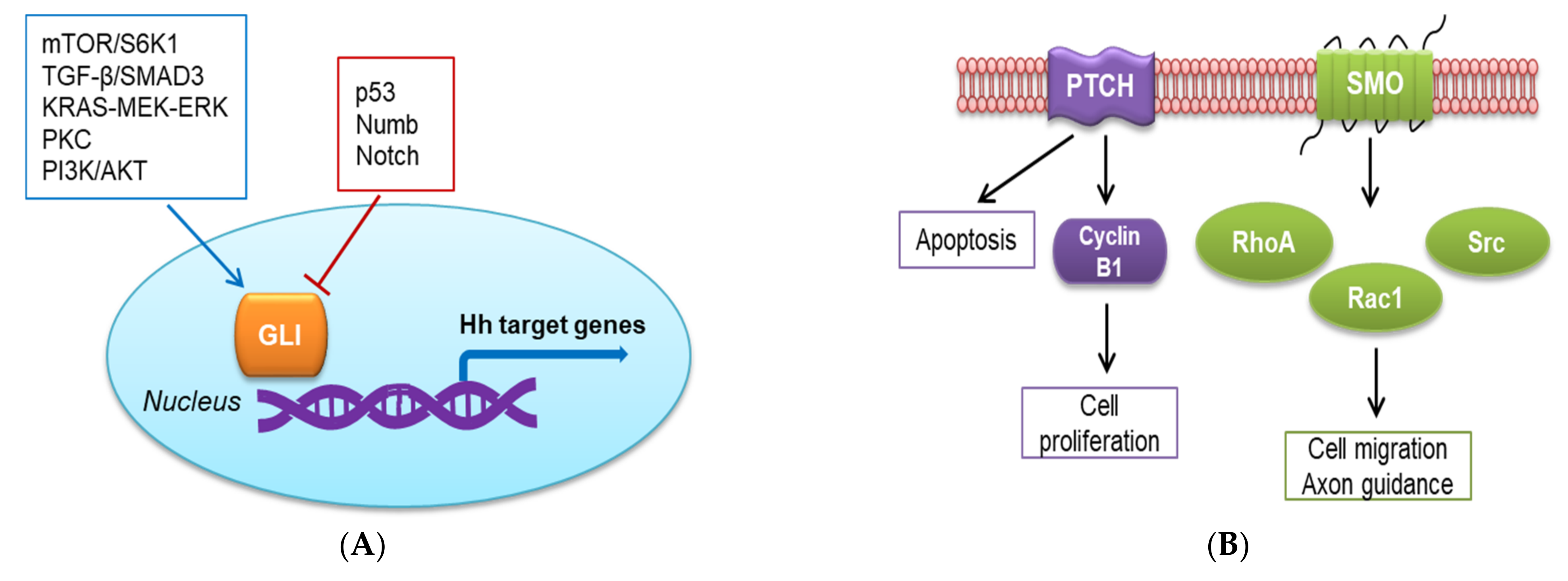
2.2. Non-Canonical Hh Signaling Pathway
3. Targeting the Hh Signaling Pathway in Cancer Therapy
3.1. Activation of the Hh Signaling Pathway in Cancer
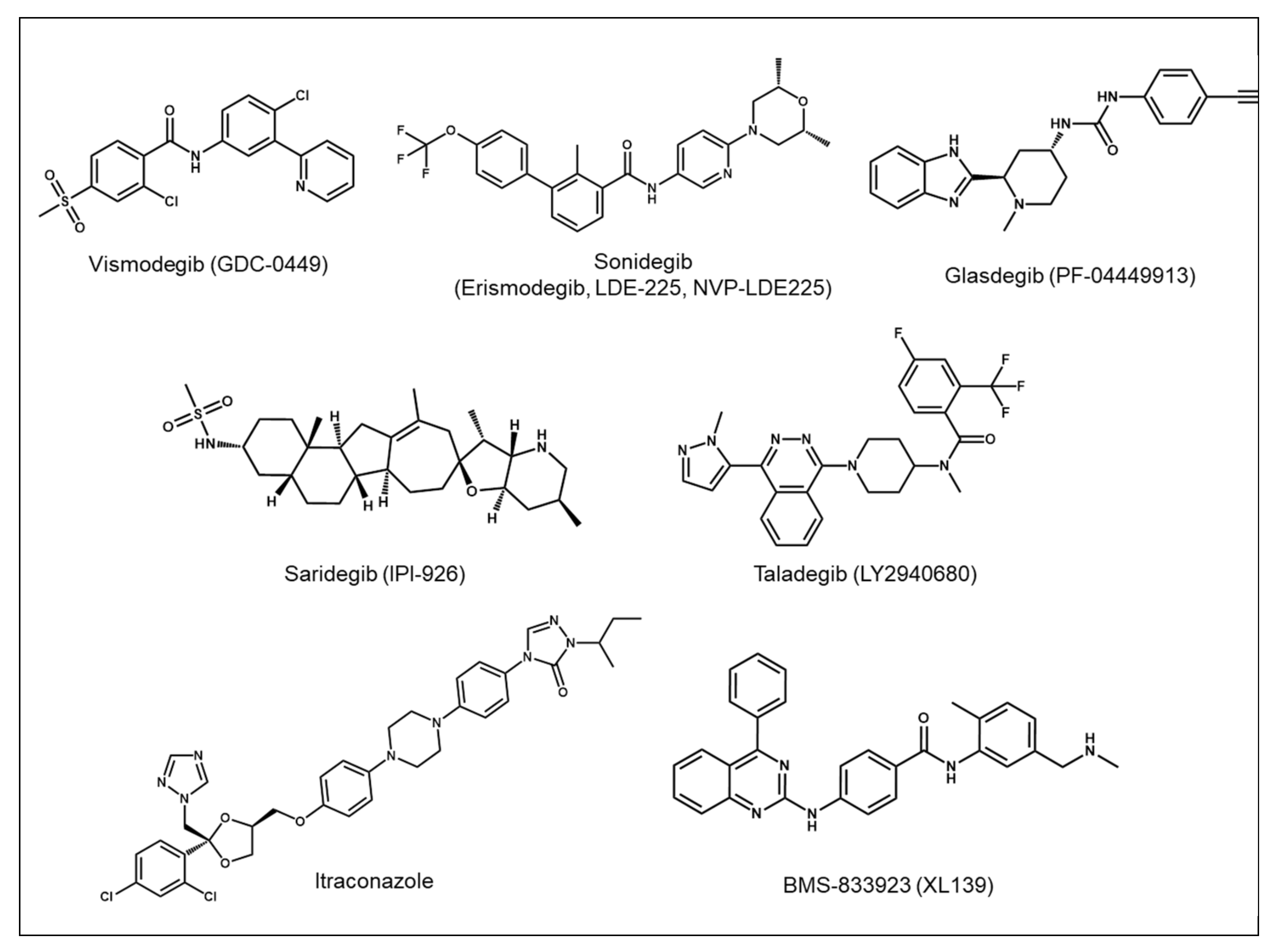
3.2. Development of Hh Pathway Inhibitors as Targeted Cancer Therapy
4. Mechanisms of Resistance to SMO Inhibitor Therapy
4.1. Development of Drug Resistance in the Clinical Context
4.2. Mechanisms of Resistance to SMO Inhibitor Therapy
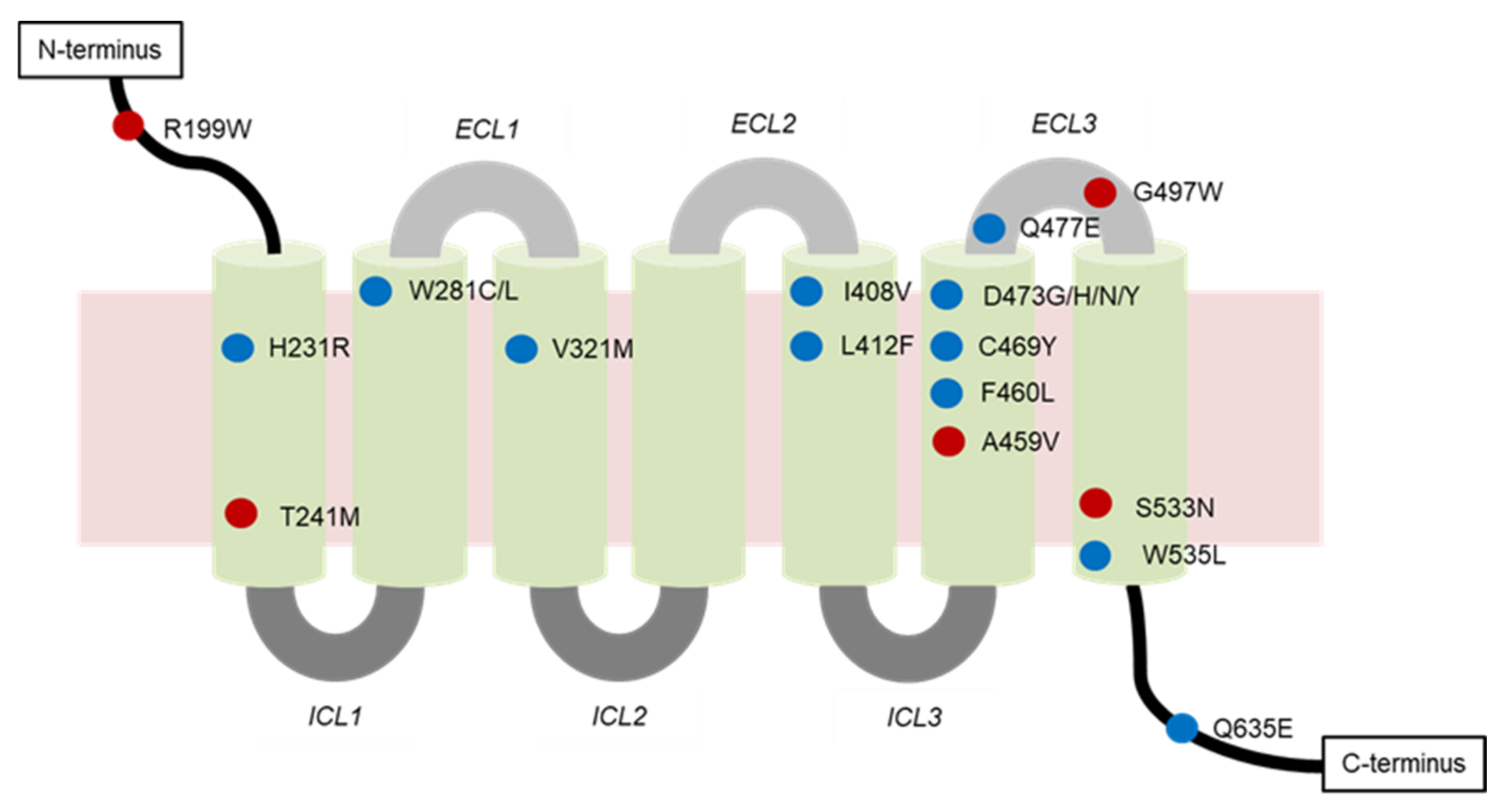
4.2.1. Genetic Mutations
- Mutations of SMO
- Mutations of Proteins other than SMO
4.2.2. Activation of the Non-Canonical Hh Pathway
4.2.3. Loss of Primary Cilia
5. Strategies to Overcome the Resistance to Hh Pathway Inhibitors
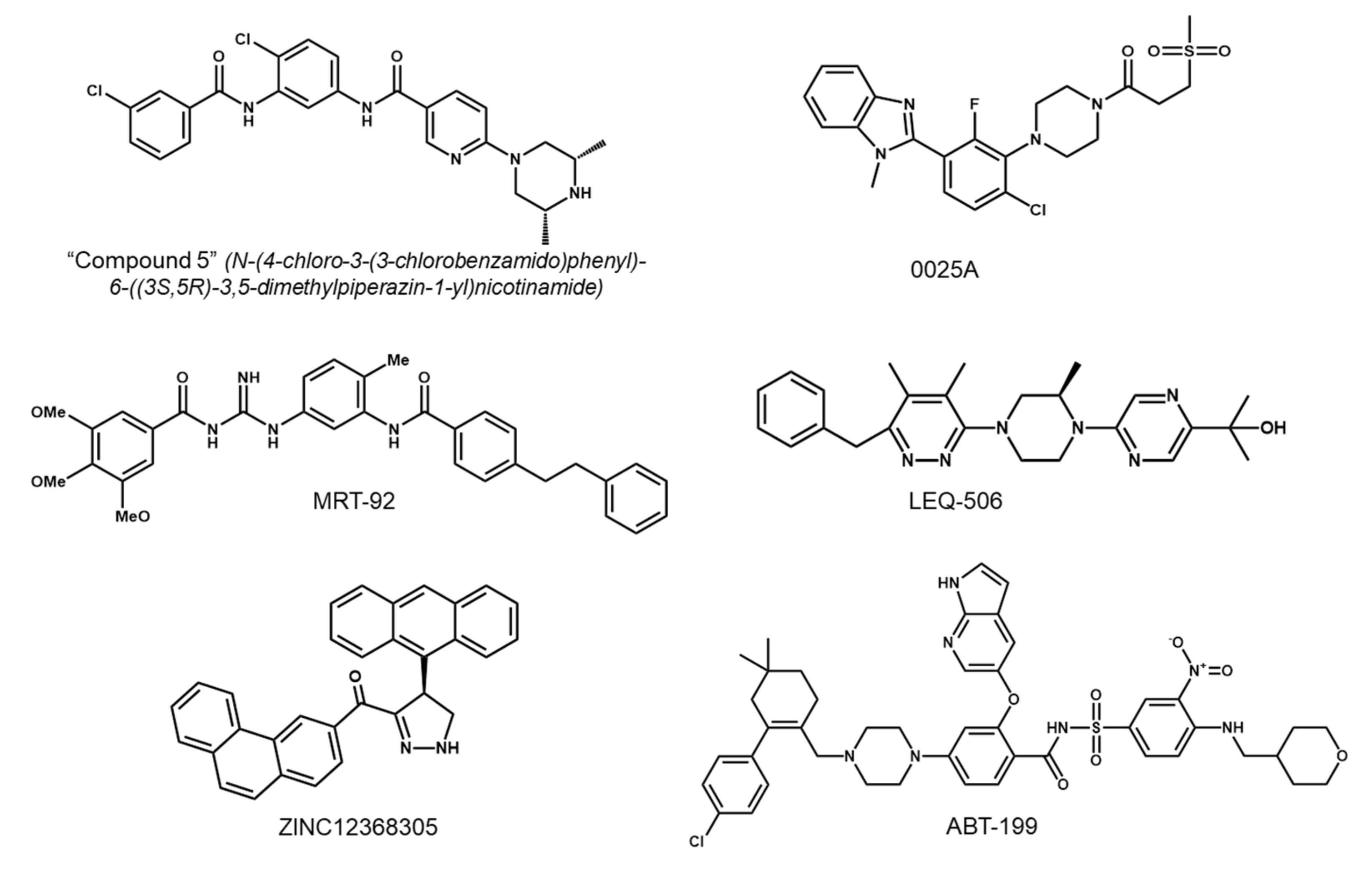
5.1. Development of Second-Generation SMO Inhibitors
5.2. Targeting Downstream Molecules of SMO
5.3. Targeting the Non-Canonical Hh Pathway
5.4. Genetic Prescreening before Initiating Cancer Therapy with Hh Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perrimon, N. Hedgehog and beyond. Cell 1995, 80, 517–520. [Google Scholar] [CrossRef]
- Ingham, P.W. Transducing Hedgehog: The story so far. EMBO J. 1998, 17, 3505–3511. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Tabin, C. The long and short of hedgehog signaling. Cell 1995, 81, 313–316. [Google Scholar] [CrossRef][Green Version]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A Cancer Stem Cell Pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Fuccillo, M.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.; Placzek, M.; Fletcher, A.G. Of mitogens and morphogens: Modelling Sonic Hedgehog mechanisms in vertebrate development. Philos. Trans. R Soc. Lond. B Biol. Sci. 2020, 375, 20190660. [Google Scholar] [CrossRef]
- Cai, C.; Thorne, J.; Grabel, L. Hedgehog Serves as a Mitogen and Survival Factor During Embryonic Stem Cell Neurogenesis. Stem Cells 2008, 26, 1097–1108. [Google Scholar] [CrossRef]
- Charron, F.; Tessier-Lavigne, M. The Hedgehog, TGF-beta/BMP and Wnt families of morphogens in axon guidance. Adv. Exp. Med. Biol. 2007, 621, 116–133. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic Medulloblastomas Contain PTCH Mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgård, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef]
- Yuan, Z.; Goetz, J.A.; Singh, S.; Ogden, S.K.; Petty, W.J.; Black, C.C.; Memoli, V.A.; Dmitrovsky, E.; Robbins, D.J. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene 2007, 26, 1046–1055. [Google Scholar] [CrossRef]
- Gupta, S.; Takebe, N.; LoRusso, P. Review: Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef]
- Danhof, R.; Lewis, K.; Brown, M. Small Molecule Inhibitors of the Hedgehog Pathway in the Treatment of Basal Cell Carcinoma of the Skin. Am. J. Clin. Dermatol. 2018, 19, 195–207. [Google Scholar] [CrossRef]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration Approval: Vismodegib for Recurrent, Locally Advanced, or Metastatic Basal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary cilia and mammalian hedgehog signaling. Cold. Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Bermúdez-Muñoz, O.M. Sonic Hedgehog (SHH) pathway in the adult brain: Key signaling for astrocyte reactivation and brain repair. Acta Biol. 2016, 38, 197–209. [Google Scholar]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Torroja, C.; Gorfinkiel, N.; Guerrero, I. Mechanisms of Hedgehog gradient formation and interpretation. J. Neurobiol. 2005, 64, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, M.; Ingham, P.W. Smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 1996, 382, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Petrov, K.; Watanabe, M.; Salic, A. Mechanism of inhibition of the tumor suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. USA 2016, 113, E5866–E5875. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Kasper, M.; Jaks, V.; Hohl, D.; Toftgård, R. Basal cell carcinoma—Molecular biology and potential new therapies. J. Clin. Investig. 2012, 122, 455–463. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with Resistance to Smoothened Antagonists by Inhibition of the PI3K Pathway in Medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front Genet. 2019, 10, 556. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of mTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1)expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal 2012, 5, re6. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS Activates Hedgehog Signaling Pathway in Pancreatic Cancer Cells*. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of Sonic Hedgehog Mediators by Transforming Growth Factor-β: Smad3-Dependent Activation of Gli2 and Gli1 Expression In vitro and In vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef]
- Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef]
- Neill, G.W.; Ghali, L.R.; Green, J.L.; Ikram, M.S.; Philpott, M.P.; Quinn, A.G. Loss of protein kinase Calpha expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 2003, 63, 4692–4697. [Google Scholar]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef]
- Marcotullio, L.D.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef]
- Dotto, G.P. Notch tumor suppressor function. Oncogene 2008, 27, 5115–5123. [Google Scholar] [CrossRef]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.-c.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi Proteins Link Hedgehog Signaling to Activation of Rho Small GTPases to Promote Fibroblast Migration*. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef]
- Yam, P.T.; Langlois, S.D.; Morin, S.; Charron, F. Sonic Hedgehog Guides Axons through a Noncanonical, Src-Family-Kinase-Dependent Signaling Pathway. Neuron 2009, 62, 349–362. [Google Scholar] [CrossRef]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Chapter three—Noncanonical Hedgehog Signaling. In Vitamins & Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 88, pp. 55–72. [Google Scholar]
- Thibert, C.; Teillet, M.-A.; Lapointe, F.; Mazelin, L.; Le Douarin, N.M.; Mehlen, P. Inhibition of Neuroepithelial Patched-Induced Apoptosis by Sonic Hedgehog. Science 2003, 301, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Thalakoti, S.; Geller, T. Chapter 8—Basal cell nevus syndrome or Gorlin syndrome. In Handbook of Clinical Neurology; Islam, M.P., Roach, E.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 132, pp. 119–128. [Google Scholar]
- Lo Muzio, L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet. J. Rare Dis. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef]
- Hutchin, M.E.; Kariapper, M.S.; Grachtchouk, M.; Wang, A.; Wei, L.; Cummings, D.; Liu, J.; Michael, L.E.; Glick, A.; Dlugosz, A.A. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: Conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005, 19, 214–223. [Google Scholar] [CrossRef]
- Abidi, A. Hedgehog signaling pathway: A novel target for cancer therapy: Vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J. Pharmacol 2014, 46, 3. [Google Scholar] [CrossRef]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differs in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.-d.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.-M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.-W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.-c.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.-G.; Toftgård, R.; Undén, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol. Med. 2009, 1, 300–302. [Google Scholar] [CrossRef]
- Szkandera, J.; Kiesslich, T.; Haybaeck, J.; Gerger, A.; Pichler, M. Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 1179–1196. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog Signaling Pathway is a New Therapeutic Target for Patients with Breast Cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef]
- Karhadkar, S.S.; Steven Bova, G.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; de Miera, E.V.-S.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals 2013, 6, 1429–1450. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Jiang, J.; Hui, C.-C. Hedgehog Signaling in Development and Cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.-W.; de Sauvage, F.J. Paracrine Hedgehog Signaling in Cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef]
- Hegde, G.V.; Peterson, K.J.; Emanuel, K.; Mittal, A.K.; Joshi, A.D.; Dickinson, J.D.; Kollessery, G.J.; Bociek, R.G.; Bierman, P.; Vose, J.M.; et al. Hedgehog-Induced Survival of B-Cell Chronic Lymphocytic Leukemia Cells in a Stromal Cell Microenvironment: A Potential New Therapeutic Target. Mol. Cancer Res. 2008, 6, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Z.; Liu, Z.; Song, C. Overcoming the emerging drug resistance of smoothened: An overview of small-molecule SMO antagonists with antiresistance activity. Future Med. Chem. 2018, 10, 2855–2875. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.T.; Zhang, R.; Boohaker, R.J. GLI1: A Therapeutic Target for Cancer. Front. Oncol. 2021, 11, 673154. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-Mediated Inhibition of Target Tissue Response to Shh Signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef]
- Sanchez, P.; Ruiz i Altaba, A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech. Dev. 2005, 122, 223–230. [Google Scholar] [CrossRef]
- Alam, M.M.; Sohoni, S.; Kalainayakan, S.P.; Garrossian, M.; Zhang, L. Cyclopamine tartrate, an inhibitor of Hedgehog signaling, strongly interferes with mitochondrial function and suppresses aerobic respiration in lung cancer cells. BMC Cancer 2016, 16, 150. [Google Scholar] [CrossRef]
- Winkler, J.D.; Isaacs, A.; Holderbaum, L.; Tatard, V.; Dahmane, N. Design and Synthesis of Inhibitors of Hedgehog Signaling Based on the Alkaloid Cyclopamine. Org. Lett. 2009, 11, 2824–2827. [Google Scholar] [CrossRef]
- Lin, T.L.; Matsui, W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. OncoTargets Ther. 2012, 5, 47. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Xie, P.; Lefrançois, P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 1089–1100.e1017. [Google Scholar] [CrossRef] [PubMed]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Richards, D.A.; Wolpin, B.M.; Becerra, C.; Hamm, J.T.; Messersmith, W.A.; Devens, S.; Cushing, J.; Goddard, J.; Schmalbach, T.; et al. The safety of IPI-926, a novel hedgehog pathway inhibitor, in combination with gemcitabine in patients (pts) with metastatic pancreatic cancer. J. Clin. Oncol. 2011, 29, 4114. [Google Scholar] [CrossRef]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef]
- Ghirga, F.; Mori, M.; Infante, P. Current trends in Hedgehog signaling pathway inhibition by small molecules. Bioorg. Med. Chem. Lett. 2018, 28, 3131–3140. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and Arsenic Trioxide Inhibit Hedgehog Pathway Activation and Tumor Growth Associated with Acquired Resistance to Smoothened Antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268. [Google Scholar] [CrossRef]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 54, 2076–2088. [Google Scholar] [CrossRef]
- Xin, M.; Ji, X.; De La Cruz, L.K.; Thareja, S.; Wang, B. Strategies to target the Hedgehog signaling pathway for cancer therapy. Med. Res. Rev. 2018, 38, 870–913. [Google Scholar] [CrossRef]
- Williams, J.A.; Guicherit, O.M.; Zaharian, B.I.; Xu, Y.; Chai, L.; Wichterle, H.; Kon, C.; Gatchalian, C.; Porter, J.A.; Rubin, L.L.; et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: Effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 4616–4621. [Google Scholar] [CrossRef]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; Kondo, S.; et al. Discovery of pyrrolo[3,2-c]quinoline-4-one derivatives as novel hedgehog signaling inhibitors. Bioorganic Med. Chem. 2012, 20, 5496–5506. [Google Scholar] [CrossRef]
- Ishii, T.; Shimizu, Y.; Nakashima, K.; Kondo, S.; Ogawa, K.; Sasaki, S.; Matsui, H. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur. J. Pharmacol. 2014, 723, 305–313. [Google Scholar] [CrossRef]
- Williams, R. Discontinued in 2013: Oncology drugs. Expert Opin. Investig. Drugs 2015, 24, 95–110. [Google Scholar] [CrossRef]
- Miller-Moslin, K.; Peukert, S.; Jain, R.K.; McEwan, M.A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; et al. 1-Amino-4-benzylphthalazines as Orally Bioavailable Smoothened Antagonists with Antitumor Activity. J. Med. Chem. 2009, 52, 3954–3968. [Google Scholar] [CrossRef]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a Second-Generation Inhibitor of Smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.-P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.-P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 4355. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Wang, W.; Hannoush, R.N.; de Sauvage, F.J. Regulation of the oncoprotein Smoothened by small molecules. Nat. Chem. Biol. 2015, 11, 246–255. [Google Scholar] [CrossRef]
- Hoch, L.; Faure, H.; Roudaut, H.; Schoenfelder, A.; Mann, A.; Girard, N.; Bihannic, L.; Ayrault, O.; Petricci, E.; Taddei, M.; et al. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor. FASEB J. 2015, 29, 1817–1829. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Oro, A.E. Initial Assessment of Tumor Regrowth After Vismodegib in Advanced Basal Cell Carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef]
- Sun, Q.; Atzmony, L.; Zaki, T.; Peng, A.; Sugarman, J.; Choate, K.A. Clues to primary vismodegib resistance lie in histology and genetics. J. Clin. Pathol. 2020, 73, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.M.; Oh, D.H.; Sanborn, J.Z.; Pincus, L.; McCalmont, T.H.; Cho, R.J. Multiple Hereditary Infundibulocystic Basal Cell Carcinoma Syndrome Associated With a Germline SUFU Mutation. JAMA Dermatol. 2016, 152, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Pricl, S.; Cortelazzi, B.; Dal Col, V.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol. Oncol. 2015, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Gerrit, J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef]
- Dijkgraaf, G.J.P.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small Molecule Inhibition of GDC-0449 Refractory Smoothened Mutants and Downstream Mechanisms of Drug Resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef]
- Gether, U.; Seifert, R.; Ballesteros, J.A.; Sanders-Bush, E.; Weinstein, H.; Kobilka, B.K. Structural Instability of a Constitutively Active G Protein-coupled Receptor: Agonist-Independent Activation Due to Conformational Flexibility. J. Biol. Chem. 1997, 272, 2587–2590. [Google Scholar] [CrossRef]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef]
- Yeh-Nayre, L.A.; Malicki, D.M.; Vinocur, D.N.; Crawford, J.R. Medulloblastoma with Excessive Nodularity: Radiographic Features and Pathologic Correlate. Case Rep. Radiol. 2012, 2012, 310359. [Google Scholar] [CrossRef]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef]
- Yao, C.D.; Haensel, D.; Gaddam, S.; Patel, T.; Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; McKellar, S.; Shankar, G.; Aasi, S.; et al. AP-1 and TGFß cooperativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat. Commun. 2020, 11, 5079. [Google Scholar] [CrossRef]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF–MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef]
- Friedman, E. Mirk/Dyrk1B in cancer. J. Cell Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 Transcriptional Activity in the Nucleus by Dyrk1*. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [PubMed]
- Zaromytidou, A.-I.; Miralles, F.; Treisman, R. MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA-binding domain. Mol. Cell Biol. 2006, 26, 4134–4148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, D.-Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.S.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway–Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Pak, E.; Ornell, K.J.; Pazyra-Murphy, M.F.; MacKenzie, E.L.; Chadwick, E.J.; Ponomaryov, T.; Kelleher, J.F.; Segal, R.A. A Transposon Screen Identifies Loss of Primary Cilia as a Mechanism of Resistance to SMO Inhibitors. Cancer Discov. 2017, 7, 1436–1449. [Google Scholar] [CrossRef]
- Fan, J.; Li, H.; Kuang, L.; Zhao, Z.; He, W.; Liu, C.; Wang, Y.; Cheng, S.Y.; Chen, W. Identification of a potent antagonist of smoothened in hedgehog signaling. Cell. Biosci. 2021, 11, 46. [Google Scholar] [CrossRef]
- Li, Q.-R.; Zhao, H.; Zhang, X.-S.; Lang, H.; Yu, K. Novel-smoothened inhibitors for therapeutic targeting of naïve and drug-resistant hedgehog pathway-driven cancers. Acta Pharmacol. Sin. 2019, 40, 257–267. [Google Scholar] [CrossRef]
- Lu, W.; Geng, D.; Sun, Z.; Yang, Z.; Ma, H.; Zheng, J.; Zhang, X. Scaffold hopping approach to a new series of smoothened antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 2300–2304. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Wu, M.; Wang, Q.; Luo, L.; Ma, H.; Zhang, X.; He, S. Discovery of a potent hedgehog pathway inhibitor capable of activating caspase8-dependent apoptosis. J. Pharmacol. Sci. 2018, 137, 256–264. [Google Scholar] [CrossRef]
- Vesci, L.; Milazzo, F.M.; Stasi, M.A.; Pace, S.; Manera, F.; Tallarico, C.; Cini, E.; Petricci, E.; Manetti, F.; De Santis, R.; et al. Hedgehog pathway inhibitors of the acylthiourea and acylguanidine class show antitumor activity on colon cancer in vitro and in vivo. Eur. J. Med. Chem. 2018, 157, 368–379. [Google Scholar] [CrossRef]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef]
- Tu, J.; Li, J.J.; Song, L.T.; Zhai, H.L.; Wang, J.; Zhang, X.Y. Molecular modeling study on resistance of WT/D473H SMO to antagonists LDE-225 and LEQ-506. Pharmacol. Res. 2018, 129, 491–499. [Google Scholar] [CrossRef]
- Sinha, N.; Chowdhury, S.; Sarkar, R.R. Molecular basis of drug resistance in smoothened receptor: An in silico study of protein resistivity and specificity. Proteins 2020, 88, 514–526. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Huang, W.-J.; Yang, J.; Tang, W.-G.; Huang, T.-M.; Tan, W.-F. ABT-199 inhibits Hedgehog pathway by acting as a competitive inhibitor of oxysterol, rather as a BH3 mimetic. Acta Pharmacol. Sin. 2021, 42, 1005–1013. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog–GLI Signaling Inhibition Suppresses Tumor Growth in Squamous Lung Cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef]
- Benvenuto, M.; Masuelli, L.; Smaele, E.D.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Stefano, E.D.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Chen, Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000, 1, 101–106. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef]
- Infante, P.; Malfanti, A.; Quaglio, D.; Balducci, S.; De Martin, S.; Bufalieri, F.; Mastrotto, F.; Basili, I.; Garofalo, M.; Lospinoso Severini, L.; et al. Glabrescione B delivery by self-assembling micelles efficiently inhibits tumor growth in preclinical models of Hedgehog-dependent medulloblastoma. Cancer Lett. 2021, 499, 220–231. [Google Scholar] [CrossRef]
- Manetti, F.; Stecca, B.; Santini, R.; Maresca, L.; Giannini, G.; Taddei, M.; Petricci, E. Pharmacophore-Based Virtual Screening for Identification of Negative Modulators of GLI1 as Potential Anticancer Agents. ACS Med. Chem. Lett. 2020, 11, 832–838. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, M.; Sun, J.; Yang, X. Casein kinase 1α: Biological mechanisms and theranostic potential. Cell Commun. Signal 2018, 16, 23. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.; Li, B.; Long, J.; Shen, C.; Yang, F.; Orton, D.; Collins, S.; Kasahara, N.; Ayad, N.G.; McCrea, H.J.; et al. A CK1α Activator Penetrates the Brain and Shows Efficacy Against Drug-resistant Metastatic Medulloblastoma. Clin. Cancer Res. 2019, 25, 1379–1388. [Google Scholar] [CrossRef]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; MacKenzie, E.L.; Zhao, X.; Pazyra-Murphy, M.F.; Park, P.M.C.; Wu, L.; Shaw, D.L.; Addleson, E.C.; Cayer, S.S.; Lopez, B.G.-C.; et al. A large-scale drug screen identifies selective inhibitors of class I HDACs as a potential therapeutic option for SHH medulloblastoma. Neuro Oncol. 2019, 21, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J. A gene transcription signature of the Akt/mTOR pathway in clinical breast tumors. Oncogene 2007, 26, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.T.; Migden, M.R.; Lewis, K.D.; Chang, A.L.S.; Guminski, A.; Gutzmer, R.; Dirix, L.; Combemale, P.; Stratigos, A.; Plummer, R.; et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef]
- Tran, D.C.; Moffat, A.; Brotherton, R.; Pague, A.; Zhu, G.A.; Chang, A.L.S. An exploratory open-label, investigator-initiated study to evaluate the efficacy and safety of combination sonidegib and buparlisib for advanced basal cell carcinomas. J. Am. Acad Dermatol. 2018, 78, 1011–1013.e1013. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SMO Inhibitor | Phase | Cancer Type | Status | ClinicalTrials.gov Identifier # |
|---|---|---|---|---|
| BMS-833923 (XL139) | Phase 1 and 2 | Chronic myeloid leukemia | Completed | NCT01218477 |
| Phase 2 | Leukemia | Terminated | NCT01357655 | |
| Glasdegib (PF-04449913) | Phase 2 | Acute myeloid leukemia | Completed | NCT01546038 |
| Phase 2 | Acute myeloid leukemia | Completed | NCT01841333 | |
| Phase 2 | Myelodysplastic syndrome Chronic myelomonocytic leukemia | Completed | NCT01842646 | |
| Phase 2 | Myelofibrosis | Terminated | NCT02226172 | |
| Phase 1 and 2 | Acute myeloid leukemia | Recruiting | NCT03390296 | |
| Phase 3 | Acute myeloid leukemia | Ongoing | NCT03416179 | |
| Phase 1 and 2 | Glioblastoma | Recruiting | NCT03466450 | |
| Phase 2 | Acute myeloid leukemia | Ongoing | NCT04051996 | |
| Phase 3 | Acute myeloid leukemia Myelodysplastic syndrome Chronic myelomonocytic leukemia | Ongoing | NCT04842604 | |
| Itraconazole | Phase 2 | Basal cell carcinoma | Completed | NCT01108094 |
| Saridegib (patidegib, IPI-926) | Phase 1 and 2 | Pancreatic cancer | Completed | NCT01130142 |
| Phase 2 | Chondrosarcoma | Completed | NCT01310816 | |
| Phase 2 | Myelofibrosis | Completed | NCT01371617 | |
| Phase 2 | Basal cell nevus syndrome | Completed | NCT02762084 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT02828111 | |
| Phase 3 | Basal cell nevus syndrome | Completed | NCT03703310 | |
| Phase 2 | Basal cell carcinoma | Terminated | NCT04155190 | |
| Phase 3 | Basal cell nevus syndrome | Ongoing | NCT04308395 | |
| Sonidegib (Erismodegib, LDE-225, NVP-LDE-225) | Phase 2 | Basal cell nevus syndrome | Completed | NCT00961896 |
| Phase 1 and 2 | Medulloblastoma | Completed | NCT01125800 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT01327053 | |
| Phase 2 | Basal cell nevus syndrome | Completed | NCT01350115 | |
| Phase 2 | Medulloblastoma | Completed | NCT01708174 | |
| Phase 2 | Breast cancer | Withdrawn | NCT01757327 | |
| Phase 1 and 2 | Myelofibrosis | Completed | NCT01787552 | |
| Phase 2 | Multiple myeloma | Ongoing | NCT02086552 | |
| Phase 2 | Multiple myeloma | Terminated | NCT02254551 | |
| Phase 2 | Basal cell carcinoma | Terminated | NCT02303041 | |
| Phase 2 and 3 | Basal cell carcinoma | Withdrawn | NCT03070691 | |
| Phase 2 | Basal cell carcinoma | Recruiting | NCT03534947 | |
| Phase 2 | Medulloblastoma | Not yet recruiting | NCT04402073 | |
| Taladegib (LY2940680) | Phase 1 and 2 | Small cell lung cancer | Terminated | NCT01722292 |
| Phase 1 and 2 | Esophageal junction cancer | Ongoing | NCT02530437 | |
| Phase 2 | Idiopathic pulmonary fibrosis | Recruiting | NCT04968574 | |
| Vismodegib (GDC-0449) | Phase 2 | Colorectal cancer | Completed | NCT00636610 |
| Phase 2 | Ovarian cancer | Completed | NCT00739661 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT00833417 | |
| Phase 2 | Small cell lung carcinoma | Completed | NCT00887159 | |
| Phase 2 | Medulloblastoma (adult) | Completed | NCT00939484 | |
| Phase 2 | Basal cell nevus syndrome | Completed | NCT00957229 | |
| Phase 2 | Ovarian cancer Basal cell carcinoma Colorectal cancer | Completed | NCT00959647 | |
| Phase 1 and 2 | Pancreatic cancer | Completed | NCT01064622 | |
| Phase 2 | Pancreatic cancer | Completed | NCT01088815 | |
| Phase 2 | Pancreatic cancer | Completed | NCT01195415 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT01201915 | |
| Phase 2 | Medulloblastoma (pediatric) | Completed | NCT01239316 | |
| Phase 2 | Chondrosarcoma | Ongoing | NCT01267955 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT01367665 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT01700049 | |
| Phase 2 | Basal cell carcinoma | Completed | NCT01815840 | |
| Phase 2 | Acute myelogenous leukemia Myelodysplastic syndrome | Terminated | NCT01880437 | |
| Phase 2 | Basal cell carcinoma | Terminated | NCT01898598 | |
| Phase 2 | B-cell lymphoma Chronic lymphocytic leukemia | Terminated | NCT01944943 | |
| Phase 2 | Solid tumors | Ongoing | NCT02091141 | |
| Phase 4 | Basal cell carcinoma | Ongoing | NCT02436408 |
| SMO Mutation | Location of Mutation | SMO Inhibitor | Type of Resistance | Cancer Type |
|---|---|---|---|---|
| A459V | Non-DBP 1 | Vismodegib | Secondary | BCC 2 |
| C469Y | DBP | Vismodegib | Secondary | BCC |
| D473G | DBP | N.D. 3 | Secondary | BCC |
| D473H | Vismodegib | Secondary | BCC, MB 4 | |
| D473H | Sonidegib | N.D. | N.D. | |
| D473N | N.D. | Primary | N.D. | |
| D473Y | Vismodegib and sonidegib | Secondary | BCC | |
| E518A | N.D. | Vismodegib | N.D. | N.D. |
| F460L | DBP | Vismodegib | N.D. | BCC |
| G497W | Non-DBP | Vismodegib | Primary | BCC |
| H231R | DBP | Vismodegib | Secondary | BCC |
| H304Y | N.D. | N.D. | N.D. | BCC |
| I408V | DBP | Vismodegib | Secondary | BCC |
| L225R | N.D. | Sonidegib | N.D. | MB |
| L412F | DBP | N.D. | Primary | BCC, meningiomas, ameloblastoma |
| N476K | N.D. | N.D. | N.D. | BCC |
| Q476 | DBP | N.D. | N.D. | BCC |
| Q477E | DBP | Vismodegib | Secondary | BCC, MB |
| Q581R | N.D. | N.D. | N.D. | BCC |
| Q635E | DBP | N.D. | N.D. | N.D. |
| R168H | N.D. | N.D. | N.D. | BCC |
| R199W | Non-DBP | N.D. | Primary | BCC |
| R302K | N.D. | N.D. | N.D. | BCC |
| S533N | Non-DBP | N.D. | Primary | BCC, primitive neuroectodermal tumors |
| T241M | Non-DBP | Vismodegib | Primary | BCC |
| V321M | DBP | Vismodegib | Secondary | BCC |
| W281C | DBP | Vismodegib | Secondary | BCC |
| W281L | N.D. | Secondary | BCC | |
| W535L | DBP | N.D. | Primary | BCC, meningiomas |
| W549X | N.D. | N.D. | N.D. | BCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.M.; Cho, J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int. J. Mol. Sci. 2022, 23, 1733. https://doi.org/10.3390/ijms23031733
Nguyen NM, Cho J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. International Journal of Molecular Sciences. 2022; 23(3):1733. https://doi.org/10.3390/ijms23031733
Chicago/Turabian StyleNguyen, Ngoc Minh, and Jungsook Cho. 2022. "Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance" International Journal of Molecular Sciences 23, no. 3: 1733. https://doi.org/10.3390/ijms23031733
APA StyleNguyen, N. M., & Cho, J. (2022). Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. International Journal of Molecular Sciences, 23(3), 1733. https://doi.org/10.3390/ijms23031733