
FAK in Cancer: From Mechanisms to Therapeutic Strategies

 ,
,  ,
,
Abstract
1. Introduction
2. Functional Regulation of FAK
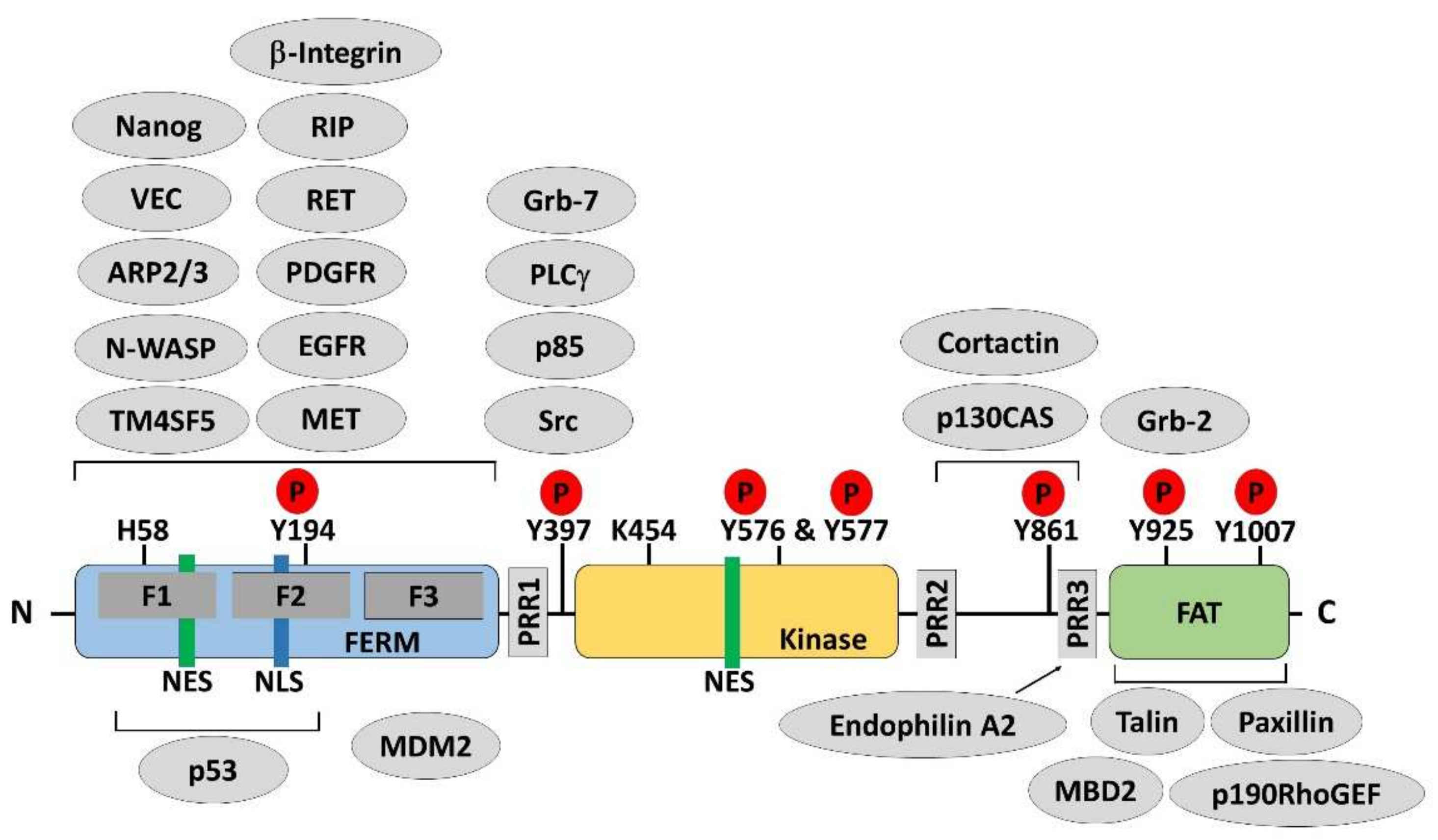
2.1. FAK Expression Regulation
2.2. FAK Activity Regulation
2.3. FAK Nuclear Translocation Regulation
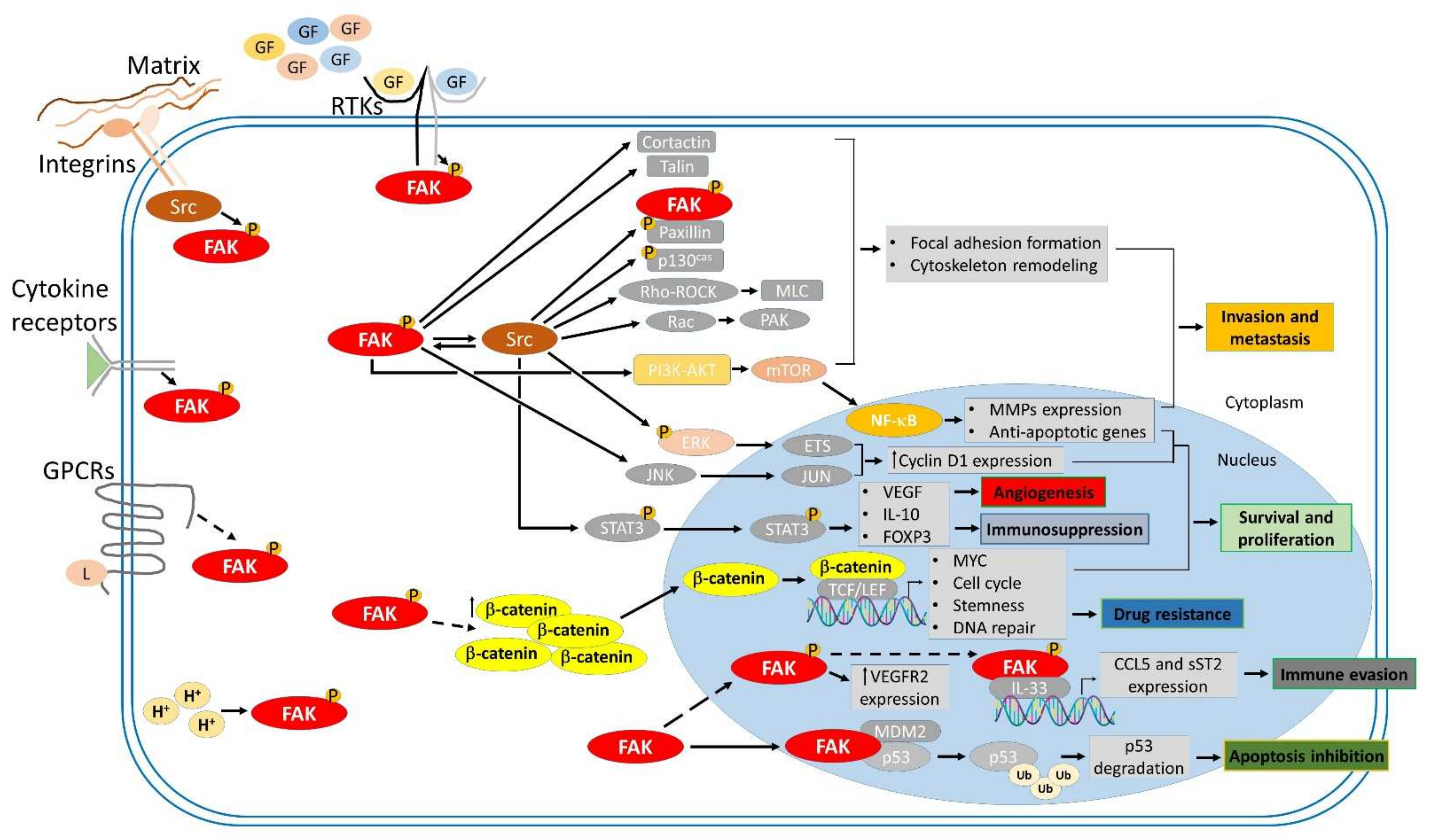
3. Roles of FAK in Cancer Progression
3.1. FAK Promotes Cell Survival and Proliferation
3.2. FAK Enhances Cancer Cell Invasion and Metastasis
3.3. FAK Tunes Mechanotransduction
3.4. FAK Drives Angiogenesis
3.5. FAK Facilitates Immunosuppressive Tumor Microenvironment
3.6. FAK Confers the Drug Resistance
4. FAK-Targeted Therapies for Cancer Treatment
4.1. FAK Enzymatic Inhibitors
4.2. FAK Scaffold Inhibitors
4.3. FAK Inhibitors in Combination Regimes
5. Conclusion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Carrera, P.M.; Kantarjian, H.M.; Blinder, V.S. The financial burden and distress of patients with cancer: Understanding and stepping-up action on the financial toxicity of cancer treatment. CA Cancer J. Clin. 2018, 68, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Calalb, M.B.; Harper, M.C.; Patel, S.K. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl. Acad. Sci. USA 1992, 89, 8487–8491. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef]
- Guan, J.L.; Trevithick, J.E.; Hynes, R.O. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul. 1991, 2, 951–964. [Google Scholar] [CrossRef]
- Kornberg, L.; Earp, H.S.; Parsons, J.T.; Schaller, M.; Juliano, R.L. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J. Biol. Chem. 1992, 267, 23439–23442. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef]
- Aboubakar Nana, F.; Vanderputten, M.; Ocak, S. Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Focal adhesion kinase as a cancer therapy target. Anti-Cancer Agents Med. Chem. 2010, 10, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Agochiya, M.; Brunton, V.G.; Owens, D.W.; Parkinson, E.K.; Paraskeva, C.; Keith, W.N.; Frame, M.C. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 1999, 18, 5646–5653. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Kaveh, F.; Baumbusch, L.O.; Nebdal, D.; Borresen-Dale, A.L.; Lingjaerde, O.C.; Edvardsen, H.; Kristensen, V.N.; Solvang, H.K. A systematic comparison of copy number alterations in four types of female cancer. BMC cancer 2016, 16, 913. [Google Scholar] [CrossRef]
- Li, C.; Bonazzoli, E.; Bellone, S.; Choi, J.; Dong, W.; Menderes, G.; Altwerger, G.; Han, C.; Manzano, A.; Bianchi, A.; et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc. Natl. Acad. Sci. USA 2019, 116, 619–624. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Kaur, A.; Cance, W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor kappa B and p53 binding sites. Biochim. Et Biophys. Acta 2004, 1678, 111–125. [Google Scholar] [CrossRef]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673. [Google Scholar] [CrossRef]
- Cheng, N.; Li, Y.; Han, Z.G. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology 2013, 57, 1906–1918. [Google Scholar] [CrossRef]
- Li, S.; Huang, X.; Zhang, D.; Huang, Q.; Pei, G.; Wang, L.; Jiang, W.; Hu, Q.; Tan, R.; Hua, Z.C. Requirement of PEA3 for transcriptional activation of FAK gene in tumor metastasis. PLoS ONE 2013, 8, e79336. [Google Scholar] [CrossRef]
- Cance, W.G.; Golubovskaya, V.M. Focal adhesion kinase versus p53: Apoptosis or survival? Sci. Signal. 2008, 1, pe22. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Finch, R.; Kweh, F.; Massoll, N.A.; Campbell-Thompson, M.; Wallace, M.R.; Cance, W.G. p53 regulates FAK expression in human tumor cells. Mol. Carcinog. 2008, 47, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.G.; Wang, Y.Y.; Fan, L.G.; Luo, H.; Han, B.; Sun, L.H.; Wang, X.F.; Zhang, J.X.; Cao, L.; Wang, X.R.; et al. MicroRNA-7 regulates glioblastoma cell invasion via targeting focal adhesion kinase expression. Chin. Med. J. 2011, 124, 2616–2621. [Google Scholar]
- Kong, X.; Li, G.; Yuan, Y.; He, Y.; Wu, X.; Zhang, W.; Wu, Z.; Chen, T.; Wu, W.; Lobie, P.E.; et al. MicroRNA-7 inhibits epithelial-to-mesenchymal transition and metastasis of breast cancer cells via targeting FAK expression. PLoS ONE 2012, 7, e41523. [Google Scholar] [CrossRef]
- Mazzu, Y.Z.; Hu, Y.; Soni, R.K.; Mojica, K.M.; Qin, L.X.; Agius, P.; Waxman, Z.M.; Mihailovic, A.; Socci, N.D.; Hendrickson, R.C.; et al. miR-193b-Regulated Signaling Networks Serve as Tumor Suppressors in Liposarcoma and Promote Adipogenesis in Adipose-Derived Stem Cells. Cancer Res. 2017, 77, 5728–5740. [Google Scholar] [CrossRef]
- Chen, J.S.; Li, H.S.; Huang, J.Q.; Dong, S.H.; Huang, Z.J.; Yi, W.; Zhan, G.F.; Feng, J.T.; Sun, J.C.; Huang, X.H. MicroRNA-379-5p inhibits tumor invasion and metastasis by targeting FAK/AKT signaling in hepatocellular carcinoma. Cancer Lett. 2016, 375, 73–83. [Google Scholar] [CrossRef]
- Bing, L.; Hong, C.; Li-Xin, S.; Wei, G. MicroRNA-543 suppresses endometrial cancer oncogenicity via targeting FAK and TWIST1 expression. Arch. Gynecol. Obstet. 2014, 290, 533–541. [Google Scholar] [CrossRef]
- Zhou, Y.; Dang, J.; Chang, K.Y.; Yau, E.; Aza-Blanc, P.; Moscat, J.; Rana, T.M. miR-1298 Inhibits Mutant KRAS-Driven Tumor Growth by Repressing FAK and LAMB3. Cancer Res. 2016, 76, 5777–5787. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Yi, J.S.; Park, H.; Lee, J.S.; Ko, Y.G. Mitsugumin 53 (MG53) ligase ubiquitinates focal adhesion kinase during skeletal myogenesis. J. Biol. Chem. 2014, 289, 3209–3216. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.P.; Fahrni, J.A.; Troie, S.; Guan, J.L.; Orth, K.; Rosen, G.D. Cleavage of focal adhesion kinase by caspases during apoptosis. J. Biol. Chem. 1997, 272, 26056–26061. [Google Scholar] [CrossRef] [PubMed]
- Crouch, D.H.; Fincham, V.J.; Frame, M.C. Targeted proteolysis of the focal adhesion kinase pp125 FAK during c-MYC-induced apoptosis is suppressed by integrin signalling. Oncogene 1996, 12, 2689–2696. [Google Scholar] [PubMed]
- Yao, L.; Li, K.; Peng, W.; Lin, Q.; Li, S.; Hu, X.; Zheng, X.; Shao, Z. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J. Transl. Med. 2014, 12, 136. [Google Scholar] [CrossRef]
- Marzia, M.; Chiusaroli, R.; Neff, L.; Kim, N.Y.; Chishti, A.H.; Baron, R.; Horne, W.C. Calpain is required for normal osteoclast function and is down-regulated by calcitonin. J. Biol. Chem. 2006, 281, 9745–9754. [Google Scholar] [CrossRef]
- Carragher, N.O.; Levkau, B.; Ross, R.; Raines, E.W. Degraded collagen fragments promote rapid disassembly of smooth muscle focal adhesions that correlates with cleavage of pp125(FAK), paxillin, and talin. J. Cell Biol. 1999, 147, 619–630. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Brami-Cherrier, K.; Gervasi, N.; Arsenieva, D.; Walkiewicz, K.; Boutterin, M.C.; Ortega, A.; Leonard, P.G.; Seantier, B.; Gasmi, L.; Bouceba, T.; et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014, 33, 356–370. [Google Scholar] [CrossRef]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Menacho, I.; Morandi, A.; Mologni, L.; Boender, P.; Gambacorti-Passerini, C.; Magee, A.I.; Hofstra, R.M.; Knowles, P.; McDonald, N.Q.; Isacke, C.M. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J. Biol. Chem. 2011, 286, 17292–17302. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.A.; Shen, T.L.; Guan, J.L. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell. Biol. 2003, 23, 8030–8041. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lietha, D.; Ceccarelli, D.F.; Karginov, A.V.; Rajfur, Z.; Jacobson, K.; Hahn, K.M.; Eck, M.J.; Schaller, M.D. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell. Biol. 2008, 28, 201–214. [Google Scholar] [CrossRef]
- Goni, G.M.; Epifano, C.; Boskovic, J.; Camacho-Artacho, M.; Zhou, J.; Bronowska, A.; Martin, M.T.; Eck, M.J.; Kremer, L.; Grater, F.; et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA 2014, 111, E3177–E3186. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Seong, J.; Tajik, A.; Sun, J.; Guan, J.L.; Humphries, M.J.; Craig, S.E.; Shekaran, A.; Garcia, A.J.; Lu, S.; Lin, M.Z.; et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 19372–19377. [Google Scholar] [CrossRef]
- Bauer, M.S.; Baumann, F.; Daday, C.; Redondo, P.; Durner, E.; Jobst, M.A.; Milles, L.F.; Mercadante, D.; Pippig, D.A.; Gaub, H.E.; et al. Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 6766–6774. [Google Scholar] [CrossRef]
- Jung, O.; Choi, S.; Jang, S.B.; Lee, S.A.; Lim, S.T.; Choi, Y.J.; Kim, H.J.; Kim, D.H.; Kwak, T.K.; Kim, H.; et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J. Cell Sci. 2012, 125, 5960–5973. [Google Scholar] [CrossRef]
- Chen, T.H.; Chan, P.C.; Chen, C.L.; Chen, H.C. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene 2011, 30, 153–166. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Daaka, Y.; Della Rocca, G.J.; Lefkowitz, R.J. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J. Biol. Chem. 1997, 272, 31648–31656. [Google Scholar] [CrossRef] [PubMed]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Goh, E.L.; Lobie, P.E. Growth hormone stimulates the tyrosine phosphorylation and association of p125 focal adhesion kinase (FAK) with JAK2. Fak is not required for stat-mediated transcription. J. Biol. Chem. 1998, 273, 10682–10689. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Webb, B.A.; Chimenti, M.S.; Jacobson, M.P.; Barber, D.L. pH sensing by FAK-His58 regulates focal adhesion remodeling. J. Cell Biol. 2013, 202, 849–859. [Google Scholar] [CrossRef] [PubMed]
- von Wichert, G.; Haimovich, B.; Feng, G.S.; Sheetz, M.P. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 2003, 22, 5023–5035. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, W.; Xia, Y.; Hawke, D.; Liu, D.X.; Lu, Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol. Cell. Biol. 2011, 31, 4258–4269. [Google Scholar] [CrossRef]
- Wu, J.C.; Chen, Y.C.; Kuo, C.T.; Wenshin Yu, H.; Chen, Y.Q.; Chiou, A.; Kuo, J.C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5, 18476. [Google Scholar] [CrossRef]
- Chuang, H.H.; Zhen, Y.Y.; Tsai, Y.C.; Chuang, C.H.; Huang, M.S.; Hsiao, M.; Yang, C.J. Targeting Pin1 for Modulation of Cell Motility and Cancer Therapy. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gomez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Griffith, B.G.C.; Upstill-Goddard, R.; Brunton, H.; Grimes, G.R.; Biankin, A.V.; Serrels, B.; Byron, A.; Frame, M.C. FAK regulates IL-33 expression by controlling chromatin accessibility at c-Jun motifs. Sci. Rep. 2021, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wu, H.J.; Guan, J.L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 2018, 8, 2550. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; McGivern, N.; Canel, M.; Byron, A.; Johnson, S.C.; McSorley, H.J.; Quinn, N.; Taggart, D.; Von Kreigsheim, A.; Anderton, S.M.; et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal. 2017, 10, eaan8355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef] [PubMed]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar] [CrossRef]
- Ossovskaya, V.; Lim, S.T.; Ota, N.; Schlaepfer, D.D.; Ilic, D. FAK nuclear export signal sequences. FEBS Lett. 2008, 582, 2402–2406. [Google Scholar] [CrossRef]
- Ahn, S.; Park, H. XIAP is essential for shear stress-enhanced Tyr-576 phosphorylation of FAK. Biochem. Biophys. Res. Commun. 2010, 399, 256–261. [Google Scholar] [CrossRef]
- Luo, S.W.; Zhang, C.; Zhang, B.; Kim, C.H.; Qiu, Y.Z.; Du, Q.S.; Mei, L.; Xiong, W.C. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 2009, 28, 2568–2582. [Google Scholar] [CrossRef]
- Kadare, G.; Toutant, M.; Formstecher, E.; Corvol, J.C.; Carnaud, M.; Boutterin, M.C.; Girault, J.A. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 2003, 278, 47434–47440. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, X.; Chen, S.; Liu, S.; Wicha, M.S.; Guan, J.L. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. 2013, 73, 5591–5602. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef]
- Xu, L.H.; Owens, L.V.; Sturge, G.C.; Yang, X.; Liu, E.T.; Craven, R.J.; Cance, W.G. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996, 7, 413–418. [Google Scholar]
- Xu, L.H.; Yang, X.; Bradham, C.A.; Brenner, D.A.; Baldwin, A.S., Jr.; Craven, R.J.; Cance, W.G. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J. Biol. Chem. 2000, 275, 30597–30604. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.D.; Xu, B.; Maschietto, M.; Marchi, F.A.; Alkailani, M.I.; Bijian, K.; Xiao, D.; Alaoui-Jamali, M.A. TRAF2 Cooperates with Focal Adhesion Signaling to Regulate Cancer Cell Susceptibility to Anoikis. Mol. Cancer Ther. 2019, 18, 139–146. [Google Scholar] [CrossRef]
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin, A.S., Jr.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef]
- Paul, R.; Luo, M.; Mo, X.; Lu, J.; Yeo, S.K.; Guan, J.L. FAK activates AKT-mTOR signaling to promote the growth and progression of MMTV-Wnt1-driven basal-like mammary tumors. Breast Cancer Res. BCR 2020, 22, 59. [Google Scholar] [CrossRef]
- Sonoda, Y.; Matsumoto, Y.; Funakoshi, M.; Yamamoto, D.; Hanks, S.K.; Kasahara, T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J. Biol. Chem. 2000, 275, 16309–16315. [Google Scholar] [CrossRef]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pestell, R.; Guan, J.L. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell 2001, 12, 4066–4077. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9. [Google Scholar] [CrossRef]
- Mugahid, D.; Kalocsay, M.; Liu, X.; Gruver, J.S.; Peshkin, L.; Kirschner, M.W. YAP regulates cell size and growth dynamics via non-cell autonomous mediators. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.H.; Yang, D.; Lim, D.S.; Wang, C.Y.; Guan, K.L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Ou, D.; Wang, Q.; Huang, Y.; Zeng, D.; Wei, T.; Ding, L.; Li, X.; Zheng, Q.; Jin, Y. Co-culture with neonatal cardiomyocytes enhances the proliferation of iPSC-derived cardiomyocytes via FAK/JNK signaling. BMC Dev. Biol. 2016, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Bosco, E.E.; Tice, D.A.; Hollingsworth, R.E.; Herbst, R.; Xiao, Z. HER2 drives Mucin-like 1 to control proliferation in breast cancer cells. Oncogene 2016, 35, 4225–4234. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Wang, P.H.; Niu, S.W.; Zhen, Y.Y.; Huang, M.S.; Hsiao, M.; Yang, C.J. Inhibition of FAK Signaling Elicits Lamin A/C-Associated Nuclear Deformity and Cellular Senescence. Front. Oncol. 2019, 9, 22. [Google Scholar] [CrossRef]
- Alza, L.; Nager, M.; Visa, A.; Canti, C.; Herreros, J. FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.E.; Lim, S.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar]
- Katoh, K. FAK-Dependent Cell Motility and Cell Elongation. Cells 2020, 9. [Google Scholar] [CrossRef]
- Horton, E.R.; Humphries, J.D.; Stutchbury, B.; Jacquemet, G.; Ballestrem, C.; Barry, S.T.; Humphries, M.J. Modulation of FAK and Src adhesion signaling occurs independently of adhesion complex composition. J. Cell Biol. 2016, 212, 349–364. [Google Scholar] [CrossRef]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; Weed, S.A. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [Google Scholar] [CrossRef]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Katoh, K. Regulation of Fibroblast Cell Polarity by Src Tyrosine Kinase. Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Lim, S.T.; Uryu, S.; Chen, X.L.; Calderwood, D.A.; Schlaepfer, D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012, 196, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Colome, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; Lopez, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Polte, T.R.; Hanks, S.K. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130(Cas)) are elevated in cytoskeleton-associated fractions following adhesion and Src transformation. Requirements for Src kinase activity and FAK proline-rich motifs. J. Biol. Chem. 1997, 272, 5501–5509. [Google Scholar] [CrossRef]
- Harte, M.T.; Hildebrand, J.D.; Burnham, M.R.; Bouton, A.H.; Parsons, J.T. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J. Biol. Chem. 1996, 271, 13649–13655. [Google Scholar] [CrossRef]
- Klemke, R.L.; Leng, J.; Molander, R.; Brooks, P.C.; Vuori, K.; Cheresh, D.A. CAS/Crk coupling serves as a "molecular switch" for induction of cell migration. J. Cell Biol. 1998, 140, 961–972. [Google Scholar] [CrossRef]
- Barfod, E.T.; Moore, A.L.; Van de Graaf, B.G.; Lidofsky, S.D. Myosin light chain kinase and Src control membrane dynamics in volume recovery from cell swelling. Mol. Biol. Cell 2011, 22, 634–650. [Google Scholar] [CrossRef]
- Chen, C.; Tao, T.; Wen, C.; He, W.Q.; Qiao, Y.N.; Gao, Y.Q.; Chen, X.; Wang, P.; Chen, C.P.; Zhao, W.; et al. Myosin light chain kinase (MLCK) regulates cell migration in a myosin regulatory light chain phosphorylation-independent mechanism. J. Biol. Chem. 2014, 289, 28478–28488. [Google Scholar] [CrossRef]
- Tornin, J.; Hermida-Prado, F.; Padda, R.S.; Gonzalez, M.V.; Alvarez-Fernandez, C.; Rey, V.; Martinez-Cruzado, L.; Estupinan, O.; Menendez, S.T.; Fernandez-Nevado, L.; et al. FUS-CHOP Promotes Invasion in Myxoid Liposarcoma through a SRC/FAK/RHO/ROCK-Dependent Pathway. Neoplasia 2018, 20, 44–56. [Google Scholar] [CrossRef]
- Kallergi, G.; Agelaki, S.; Markomanolaki, H.; Georgoulias, V.; Stournaras, C. Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell Physiol. Biochem. 2007, 20, 977–986. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H. Adhesion signaling - crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Schlaepfer, D.D. Focal adhesion kinase: Switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 2009, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Lawson, C.; Ghassemian, M.; Schlaepfer, D.D. Cortactin as a target for FAK in the regulation of focal adhesion dynamics. PLoS ONE 2012, 7, e44041. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Suetsugu, S.; Cooper, L.A.; Takenawa, T.; Guan, J.L. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 2004, 279, 9565–9576. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, V.; Fischer, R.S.; Waterman, C.M. The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell 2016, 27, 1085–1100. [Google Scholar] [CrossRef]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef]
- Tang, H.; Li, A.; Bi, J.; Veltman, D.M.; Zech, T.; Spence, H.J.; Yu, X.; Timpson, P.; Insall, R.H.; Frame, M.C.; et al. Loss of Scar/WAVE complex promotes N-WASP- and FAK-dependent invasion. Curr. Biol. 2013, 23, 107–117. [Google Scholar] [CrossRef]
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999, 274, 24425–24430. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef]
- Huang, K.; Gao, N.; Bian, D.; Zhai, Q.; Yang, P.; Li, M.; Wang, X. Correlation between FAK and EGF-Induced EMT in Colorectal Cancer Cells. J. Oncol 2020, 2020, 5428920. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757–4772. [Google Scholar] [CrossRef]
- Zheng, D.; Duan, H.; Wang, S.; Xu, Q.; Gan, L.; Li, J.; Dong, Q. FAK regulates epithelialmesenchymal transition in adenomyosis. Mol. Med. Rep. 2018, 18, 5461–5472. [Google Scholar] [CrossRef]
- Li, J.; Hao, N.; Han, J.; Zhang, M.; Li, X.; Yang, N. ZKSCAN3 drives tumor metastasis via integrin beta4/FAK/AKT mediated epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Cell Int. 2020, 20, 216. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhou, X.; Rowe, R.G.; Hu, Y.; Schlaepfer, D.D.; Ilic, D.; Dressler, G.; Park, A.; Guan, J.L.; Weiss, S.J. Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J. Cell Biol. 2011, 195, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Tai, H.C.; Lin, T.H.; Wang, S.W.; Lin, C.Y.; Chao, C.C.; Yu, H.J.; Tsai, Y.C.; Lai, Y.W.; Lin, C.W.; et al. CCN3 promotes epithelial-mesenchymal transition in prostate cancer via FAK/Akt/HIF-1alpha-induced twist expression. Oncotarget 2017, 8, 74506–74518. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent beta1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Wyke, A.W.; Jones, R.J.; McLean, G.W.; Westhoff, M.A.; Brunton, V.G.; Frame, M.C. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol. 2002, 4, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gan, B.; Yoo, Y.; Guan, J.L. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev. Cell 2005, 9, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef]
- Lu, H.; Hu, L.; Yu, L.; Wang, X.; Urvalek, A.M.; Li, T.; Shen, C.; Mukherjee, D.; Lahiri, S.K.; Wason, M.S.; et al. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene 2014, 33, 2909–2917. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1beta activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef]
- Chin, L.; Xia, Y.; Discher, D.E.; Janmey, P.A. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Zhou, D.W.; Lee, T.T.; Weng, S.; Fu, J.; Garcia, A.J. Effects of substrate stiffness and actomyosin contractility on coupling between force transmission and vinculin-paxillin recruitment at single focal adhesions. Mol. Biol. Cell 2017, 28, 1901–1911. [Google Scholar] [CrossRef]
- Bell, S.; Terentjev, E.M. Focal Adhesion Kinase: The Reversible Molecular Mechanosensor. Biophys. J. 2017, 112, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.F.; Li, Q.; Melnichouk, O.; Huszti, E.; Martin, L.J.; Gunasekara, A.; Mawdsley, G.; Yaffe, M.J.; Minkin, S. Evidence that breast tissue stiffness is associated with risk of breast cancer. PLoS ONE 2014, 9, e100937. [Google Scholar] [CrossRef] [PubMed]
- Rens, E.G.; Merks, R.M.H. Cell Shape and Durotaxis Explained from Cell-Extracellular Matrix Forces and Focal Adhesion Dynamics. iScience 2020, 23, 101488. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Ling, J.Y.; Chen, W.C.; Lin, H.H.; Tang, M.J. Mechanotransduction of matrix stiffness in regulation of focal adhesion size and number: Reciprocal regulation of caveolin-1 and beta1 integrin. Sci. Rep. 2017, 7, 15008. [Google Scholar] [CrossRef] [PubMed]
- Radel, C.; Rizzo, V. Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H936–H945. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grande-Garcia, A.; Echarri, A.; de Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; del Pozo, M.A. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vicente, R.; Pavon, D.M.; Martin-Padura, I.; Catala-Montoro, M.; Diez-Sanchez, A.; Quilez-Alvarez, A.; Lopez, J.A.; Sanchez-Alvarez, M.; Vazquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, Z.; Chen, Y.; Li, S.; Jiang, Y.; Yang, H.; Wu, C.; You, F.; Zheng, C.; Zhu, J.; et al. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta Biomater. 2019, 88, 86–101. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Rio Hernandez, A.E. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skladal, P.; Pesl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [PubMed]
- Wullkopf, L.; West, A.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef]
- Gehler, S.; Compere, F.V.; Miller, A.M. Semaphorin 3A Increases FAK Phosphorylation at Focal Adhesions to Modulate MDA-MB-231 Cell Migration and Spreading on Different Substratum Concentrations. Int. J. Breast Cancer 2017, 2017, 9619734. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef]
- Allen, S.C.; Widman, J.A.; Datta, A.; Suggs, L.J. Dynamic extracellular matrix stiffening induces a phenotypic transformation and a migratory shift in epithelial cells. Integr. Biol. 2020, 12, 161–174. [Google Scholar] [CrossRef]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef]
- Januszyk, M.; Kwon, S.H.; Wong, V.W.; Padmanabhan, J.; Maan, Z.N.; Whittam, A.J.; Major, M.R.; Gurtner, G.C. The Role of Focal Adhesion Kinase in Keratinocyte Fibrogenic Gene Expression. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Lechertier, T.; Hodivala-Dilke, K. Focal adhesion kinase and tumour angiogenesis. J. Pathol. 2012, 226, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Tomar, A.; Miller, N.L.; Yoo, J.; Schlaepfer, D.D. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J. Biol. Chem. 2010, 285, 21526–21536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Peng, X.; Sun, S.; Park, A.Y.; Guan, J.L. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J. Cell Biol. 2010, 189, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.L.; Park, A.Y.; Alcaraz, A.; Peng, X.; Jang, I.; Koni, P.; Flavell, R.A.; Gu, H.; Guan, J.L. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell Biol. 2005, 169, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Braren, R.; Hu, H.; Kim, Y.H.; Beggs, H.E.; Reichardt, L.F.; Wang, R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J. Cell Biol. 2006, 172, 151–162. [Google Scholar] [CrossRef]
- Shiau, J.-P.; Wu, C.-C.; Chang, S.-J.; Pan, M.-R.; Liu, W.; Ou-Yang, F.; Chen, F.-M.; Hou, M.-F.; Shih, S.-L.; Luo, C.-W. FAK Regulates VEGFR2 Expression and Promotes Angiogenesis in Triple-Negative Breast Cancer. Biomedicines 2021, 9, 1789. [Google Scholar] [CrossRef]
- Cheng, S.; Zhang, X.; Xu, Y.; Dai, X.; Li, J.; Zhang, T.; Chen, X. Kruppel-like factor 8 regulates VEGFA expression and angiogenesis in hepatocellular carcinoma. Sci. Rep. 2018, 8, 17415. [Google Scholar] [CrossRef]
- Schmidt, T.T.; Tauseef, M.; Yue, L.; Bonini, M.G.; Gothert, J.; Shen, T.L.; Guan, J.L.; Predescu, S.; Sadikot, R.; Mehta, D. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L291–L300. [Google Scholar] [CrossRef]
- Tavora, B.; Batista, S.; Reynolds, L.E.; Jadeja, S.; Robinson, S.; Kostourou, V.; Hart, I.; Fruttiger, M.; Parsons, M.; Hodivala-Dilke, K.M. Endothelial FAK is required for tumour angiogenesis. EMBO Mol. Med. 2010, 2, 516–528. [Google Scholar] [CrossRef]
- Lu, C.; Bonome, T.; Li, Y.; Kamat, A.A.; Han, L.Y.; Schmandt, R.; Coleman, R.L.; Gershenson, D.M.; Jaffe, R.B.; Birrer, M.J.; et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007, 67, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.; Chen, X.L.; Nam, J.O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.; Ghassemian, M.; et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, A.R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, A.; Bologna, M. Targeting vascular cell migration as a strategy for blocking angiogenesis: The central role of focal adhesion protein tyrosine kinase family. Curr. Pharm. Des. 2007, 13, 2129–2145. [Google Scholar] [CrossRef]
- Cabrita, M.A.; Jones, L.M.; Quizi, J.L.; Sabourin, L.A.; McKay, B.C.; Addison, C.L. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol. Oncol. 2011, 5, 517–526. [Google Scholar] [CrossRef]
- Ward, K.K.; Tancioni, I.; Lawson, C.; Miller, N.L.; Jean, C.; Chen, X.L.; Uryu, S.; Kim, J.; Tarin, D.; Stupack, D.G.; et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin. Exp. Metastasis 2013, 30, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Zhou, S.; Yu, D.; Gu, J.; Qin, Q.; Cheng, Y.; Sun, X. Focal adhesion kinase: Insight into its roles and therapeutic potential in oesophageal cancer. Cancer Lett. 2021, 496, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Liang, S.M.; Wu, Y.J.; Wu, Y.J.; Lu, Y.J.; Jan, Y.J.; Ko, B.S.; Chuang, Y.J.; Shyue, S.K.; Kuo, C.C.; et al. Cordycepin Suppresses Endothelial Cell Proliferation, Migration, Angiogenesis, and Tumor Growth by Regulating Focal Adhesion Kinase and p53. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Laszlo, V.; Valko, Z.; Ozsvar, J.; Kovacs, I.; Garay, T.; Hoda, M.A.; Klikovits, T.; Stockhammer, P.; Aigner, C.; Groger, M.; et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J. Mol. Med. 2019, 97, 231–242. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Lechertier, T.; Reynolds, L.E.; Kim, H.; Pedrosa, A.R.; Gomez-Escudero, J.; Munoz-Felix, J.M.; Batista, S.; Dukinfield, M.; Demircioglu, F.; Wong, P.P.; et al. Pericyte FAK negatively regulates Gas6/Axl signalling to suppress tumour angiogenesis and tumour growth. Nat. Commun. 2020, 11, 2810. [Google Scholar] [CrossRef] [PubMed]
- Lees, D.M.; Reynolds, L.E.; Pedrosa, A.R.; Roy-Luzarraga, M.; Hodivala-Dilke, K.M. Phosphorylation of pericyte FAK-Y861 affects tumour cell apoptosis and tumour blood vessel regression. Angiogenesis 2021, 24, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Minasian, L.; Kohn, E.C. New strategies in ovarian cancer treatment. Cancer 2019, 125 (Suppl. 24), 4623–4629. [Google Scholar] [CrossRef] [PubMed]
- Diaz Osterman, C.J.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 2019, 8. [Google Scholar] [CrossRef]
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 2013, 105, 1485–1495. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Gao, C.; Chen, G.; Kuan, S.F.; Zhang, D.H.; Schlaepfer, D.D.; Hu, J. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife 2015, 4. [Google Scholar] [CrossRef]
- Teeuwssen, M.; Fodde, R. Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell-cell adhesion: Linking Wnt/beta-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, J.; Wang, L.; Xiao, S.; Li, L.; Yan, X.; Yao, W.; Wu, L.; Zhang, S.; Zhang, Y.; et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cancer Lett. 2019, 450, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sun, Q.; Li, X.; Feng, J.; Ao, Z.; Li, X.; Wang, J. Substrate Stiffness Modulates the Growth, Phenotype, and Chemoresistance of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 718834. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Chen, G.; Gao, C.; Gao, X.; Zhang, D.H.; Kuan, S.F.; Burns, T.F.; Hu, J. Wnt/beta-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 806–813. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef]
- Roy-Luzarraga, M.; Hodivala-Dilke, K. Molecular Pathways: Endothelial Cell FAK-A Target for Cancer Treatment. Clin. Cancer Res. 2016, 22, 3718–3724. [Google Scholar] [CrossRef]
- Eke, I.; Deuse, Y.; Hehlgans, S.; Gurtner, K.; Krause, M.; Baumann, M.; Shevchenko, A.; Sandfort, V.; Cordes, N. β1Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Investig. 2012, 122, 1529–1540. [Google Scholar] [CrossRef]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef]
- Tang, K.J.; Constanzo, J.D.; Venkateswaran, N.; Melegari, M.; Ilcheva, M.; Morales, J.C.; Skoulidis, F.; Heymach, J.V.; Boothman, D.A.; Scaglioni, P.P. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin. Cancer Res. 2016, 22, 5851–5863. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.D.; Giri, U.; Yang, L.; Woo, S.H.; Story, M.D.; Pickering, C.R.; Byers, L.A.; Williams, M.D.; El-Naggar, A.; Wang, J.; et al. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clin. Cancer Res. 2016, 22, 4643–4650. [Google Scholar] [CrossRef] [PubMed]
- Hirt, U.A.; Waizenegger, I.C.; Schweifer, N.; Haslinger, C.; Gerlach, D.; Braunger, J.; Weyer-Czernilofsky, U.; Stadtmuller, H.; Sapountzis, I.; Bader, G.; et al. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Hafliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, D.H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Lim, S.T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Gothert, J.R.; Shen, T.L.; Guan, J.L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 2008, 181, 43–50. [Google Scholar] [CrossRef]
- Han, S.; Mistry, A.; Chang, J.S.; Cunningham, D.; Griffor, M.; Bonnette, P.C.; Wang, H.; Chrunyk, B.A.; Aspnes, G.E.; Walker, D.P.; et al. Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J. Biol. Chem. 2009, 284, 13193–13201. [Google Scholar] [CrossRef]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol. Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Virnig, C.; Cance, W.G. TAE226-induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol. Carcinog. 2008, 47, 222–234. [Google Scholar] [CrossRef]
- Liu, T.J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Seenisamy, J.; Emmanuvel, L.; Kulkarni, S.S.; Bomke, J.; Rohdich, F.; Greiner, H.; Esdar, C.; Krier, M.; Gradler, U.; et al. Fragment-based discovery of new highly substituted 1H-pyrrolo[2,3-b]- and 3H-imidazolo[4,5-b]-pyridines as focal adhesion kinase inhibitors. J. Med. Chem. 2013, 56, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Hayashi, Y.; Suzuki, S.; Oomori, Y.; Aramaki, Y.; Matsushita, Y.; Iwatani, M.; Iwata, H.; Okabe, A.; Awazu, Y.; et al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg Med Chem Lett. 2013, 23, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.V.; Hunt, D.L.; He, D.; Magis, A.T.; Ostrov, D.A.; Cance, W.G. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 2009, 52, 4716–4724. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC cancer 2013, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo[3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shao, H.; Golubovskaya, V.M.; Chen, H.; Cance, W.; Adjei, A.A.; Dy, G.K. Efficacy of focal adhesion kinase inhibition in non-small cell lung cancer with oncogenically activated MAPK pathways. Br. J. Cancer 2016, 115, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Halder, J.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Honda, T.; Kamat, A.A.; Han, L.Y.; Kim, T.J.; Lu, C.; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007, 67, 10976–10983. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 77, 997–1003. [Google Scholar] [CrossRef]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Antoniades, I.; Kyriakou, M.; Charalambous, A.; Kalalidou, K.; Christodoulou, A.; Christoforou, M.; Skourides, P.A. FAK displacement from focal adhesions: A promising strategy to target processes implicated in cancer progression and metastasis. Cell Commun. Signal. 2021, 19, 3. [Google Scholar] [CrossRef]
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Seto, T.; Kato, T.; Nishio, M.; Goto, K.; Atagi, S.; Hosomi, Y.; Yamamoto, N.; Hida, T.; Maemondo, M.; Nakagawa, K.; et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): An open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014, 15, 1236–1244. [Google Scholar] [CrossRef]
- Kitagawa, C.; Mori, M.; Ichiki, M.; Sukoh, N.; Kada, A.; Saito, A.M.; Ichinose, Y. Gefitinib Plus Bevacizumab vs. Gefitinib Alone for EGFR Mutant Non-squamous Non-small Cell Lung Cancer. In Vivo 2019, 33, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Hsu, P.C.; Huang, C.Y.; Wang, C.C.; Kuo, S.C.; Chu, C.H.; Tung, P.H.; Huang, A.C.; Wang, C.L.; Chiu, L.C.; Fang, Y.F.; et al. The Combination of Afatinib and Bevacizumab in Untreated EGFR-Mutated Advanced Lung Adenocarcinoma: A Multicenter Observational Study. Pharmaceuticals 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Paz-Ares, L.G.; Yang, J.C.; Lee, K.H.; Garrido, P.; Park, K.; Kim, J.H.; Lee, D.H.; Mao, H.; Wijayawardana, S.R.; et al. Phase I Study of the Efficacy and Safety of Ramucirumab in Combination with Osimertinib in Advanced T790M-positive EGFR-mutant Non-small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 992–1002. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Molecular Targets | Type | Cancer Types | References |
|---|---|---|---|---|
| BI-853520 (IN10018) | FAK | KI | Prostate cancer; breast cancer | [213,214] |
| GSK2256098 | FAK | KI | Pancreatic cancer; ovarian cancer | [215,216] |
| NVP-TAC544 | FAK | KI | N/A | [217] |
| PF-431396 | FAK/PYK2 | KI | Pancreatic cancer; pleural mesothelioma | [218,219] |
| PF-573228 | FAK | KI | Pancreatic cancer; pleural mesothelioma; lung cancer | [89,219,220] |
| TAE226 | FAK/IGF-IR | KI | Breast cancer; ovarian carcinoma; glioma | [221,222] |
| VS-4718 (PND-1186) | FAK/PYK2 | KI | Breast cancer/ovarian cancer; pancreatic cancers | [191,223] |
| VS-6062 (PF-00562271) | FAK/PYK2 | KI | Gliomas; pancreatic cancer; colon cancer; lung cancer; prostate cancer; breast cancer | [224,225,226] |
| VS-6063 (Defactinib) | FAK/PYK2 | KI | Ovarian cancer | [195] |
| 1H-Pyrrolo(2,3-b) pyridine | N/A | aKI | N/A | [227] |
| Compound 1 and 2 | N/A | aKI | N/A | [228] |
| C4 | FAK-VEGFR3 interaction | SI | Breast cancer | [229] |
| R2 | FAK-p53 interaction | SI | Colorectal cancer | [230] |
| Y11 | FAK | SI | Colon cancer and breast cancer | [231] |
| Y15 | FAK | SI | Breast cancer; lung cancer | [232,233] |
| Drugs | Molecular Targets | Type | Cancer Types | Trial Identifiers |
|---|---|---|---|---|
| APG-2449 | Multiple kinases | KI | Advanced Solid Cancer | NCT03917043 (I) |
| BI-853520 (IN10018) | FAK | KI | Metastatic Nonhematologic Malignancies | NCT01335269 (I) |
| Defactinib (VS-6063) | FAK/PYK2 | KI | Nonhematologic Malignancies; Lung Cancer; Malignant Pleural Mesothelioma | NCT00787033 (I) NCT01943292 (I) NCT01951690 (II) NCT02004028 (II) |
| Defactinib (VS-6063) Avelumab | FAK/PYK2 PD-L1 | KI | Epithelial Ovarian Cancer | NCT02943317 (I) |
| Defactinib (VS-6063) Paclitaxel | FAK/PYK2 Tubulin | KI | Ovarian Cancer | NCT01778803 (I) |
| Defactinib (VS-6063) Paclitaxel Carboplatin | FAK/PYK2 Tubulin DNA | KI | Ovarian Cancer | NCT03287271 (I/II) |
| Defactinib (VS-6063) Pembrolizumab | FAK/PYK2 PD-1 | KI | Pancreatic Ductal Adenocarcinoma; Advanced Solid Malignancies | NCT02758587 (I/II) NCT03727880 (II) |
| Defactinib (VS-6063) Pembrolizumab Gemcitabine | FAK/PYK2 PD-1 DNA | KI | Advanced Solid Tumors; Pancreatic Cancer | NCT02546531 (I) |
| Defactinib (VS-6063) RO5126766 | FAK/PYK2 RAF/MEK | KI | NSCLC; Solid Tumor; Low-Grade Serous Ovarian Cancer; Colorectal Cancer | NCT03875820 (I) |
| Defactinib (VS-6063) VS-5584 | FAK/PYK2 PI3K/mTOR | KI | Relapsed Malignant Mesothelioma | NCT02372227 (I) |
| Defactinib (VS-6063) VS-6766 | FAK/PYK2 RAF/MEK | KI | Ovarian Cancer; Metastatic Uveal Melanoma; Non-Small Cell Lung Cancer with KRAS Activating Mutation | NCT04620330 (II) NCT04625270 (II) NCT04720417 (II) |
| GSK2256098 | FAK | KI | Solid Tumors | NCT00996671 (I) NCT01138033 (I) |
| GSK2256098 Trametinib | FAK MEK | KI | Advanced Solid Cancer | NCT01938443 (I) |
| GSK2256098 Vismodegib Capivasertib Abemaciclib | FAK Smoothened receptor AKT CDK4/CDK6 | KI | Progressive Meningiomas | NCT02523014 (II) |
| VS-4718 (PND-1186) | FAK/PYK2 | KI | Metastatic Nonhematologic Cancers; Acute Myeloid or B-Cell Acute Lymphoblastic Leukemia | NCT01849744 (I) NCT02215629 (I) |
| VS-4718 (PND-1186) Nab-paclitaxel Gemcitabine | FAK/PYK2 Tubulin DNA | KI | Pancreatic Cancer | NCT02651727 (I) |
| VS-6062 (PF-00562271) | FAK/PYK2 | KI | Pancreatic, Head and Neck, Prostatic Neoplasms | NCT00666926 (I) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, H.-H.; Zhen, Y.-Y.; Tsai, Y.-C.; Chuang, C.-H.; Hsiao, M.; Huang, M.-S.; Yang, C.-J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. https://doi.org/10.3390/ijms23031726
Chuang H-H, Zhen Y-Y, Tsai Y-C, Chuang C-H, Hsiao M, Huang M-S, Yang C-J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. International Journal of Molecular Sciences. 2022; 23(3):1726. https://doi.org/10.3390/ijms23031726
Chicago/Turabian StyleChuang, Hsiang-Hao, Yen-Yi Zhen, Yu-Chen Tsai, Cheng-Hao Chuang, Michael Hsiao, Ming-Shyan Huang, and Chih-Jen Yang. 2022. "FAK in Cancer: From Mechanisms to Therapeutic Strategies" International Journal of Molecular Sciences 23, no. 3: 1726. https://doi.org/10.3390/ijms23031726
APA StyleChuang, H.-H., Zhen, Y.-Y., Tsai, Y.-C., Chuang, C.-H., Hsiao, M., Huang, M.-S., & Yang, C.-J. (2022). FAK in Cancer: From Mechanisms to Therapeutic Strategies. International Journal of Molecular Sciences, 23(3), 1726. https://doi.org/10.3390/ijms23031726