
DNA Polymerase-Parental DNA Interaction Is Essential for Helicase-Polymerase Coupling during Bacteriophage T7 DNA Replication


Abstract
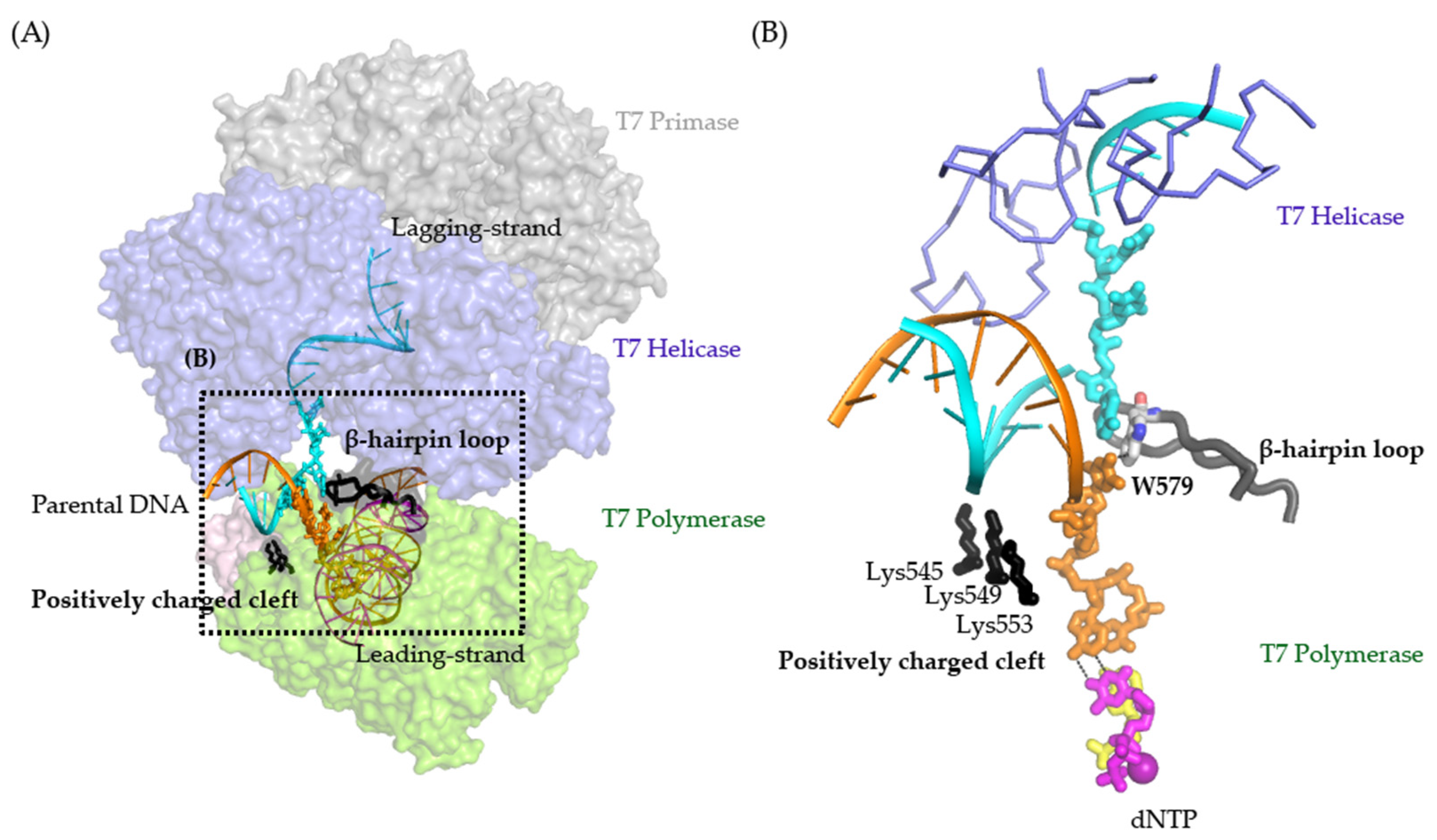
:1. Introduction
2. Results
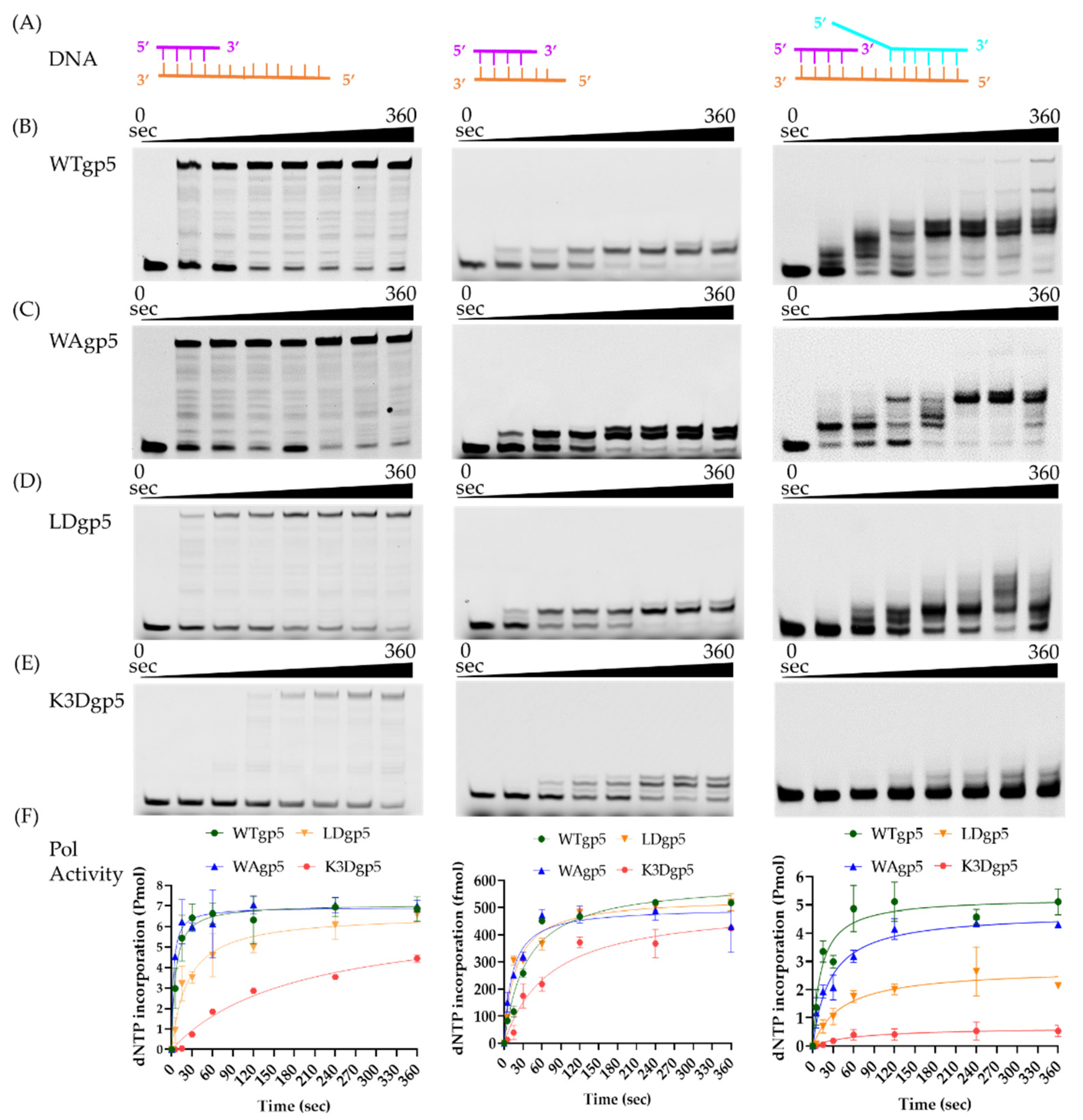
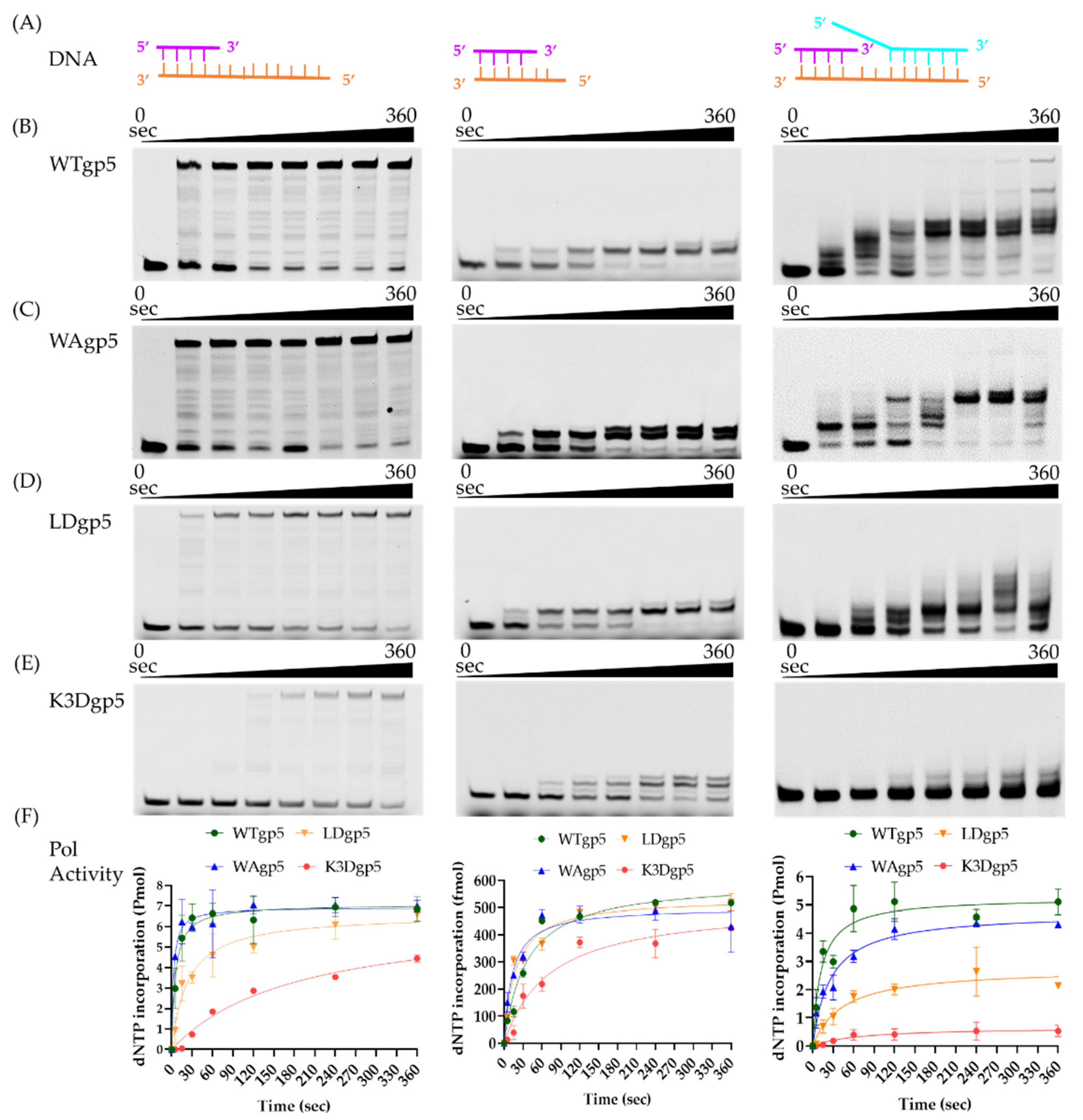
2.1. DNA Synthesis by Wild-Type (WT) and Mutant gp5-trx
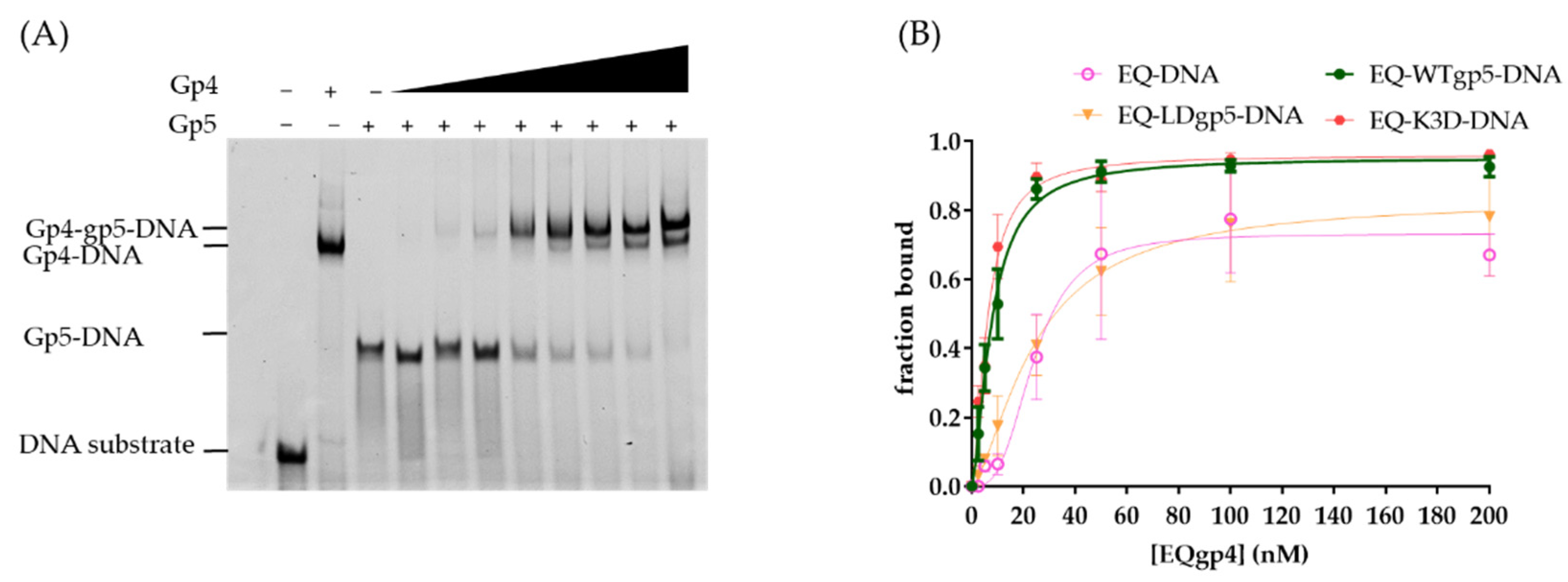
2.2. DNA Binding by WT and Mutant gp5-trx
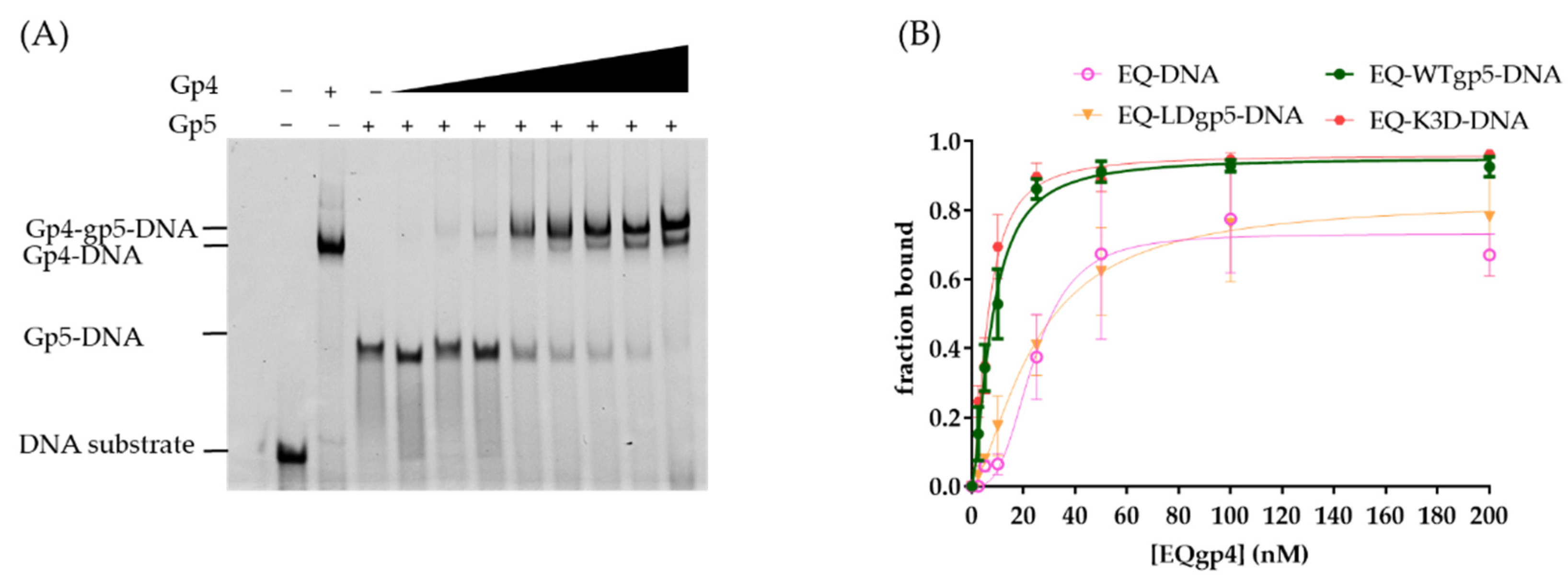
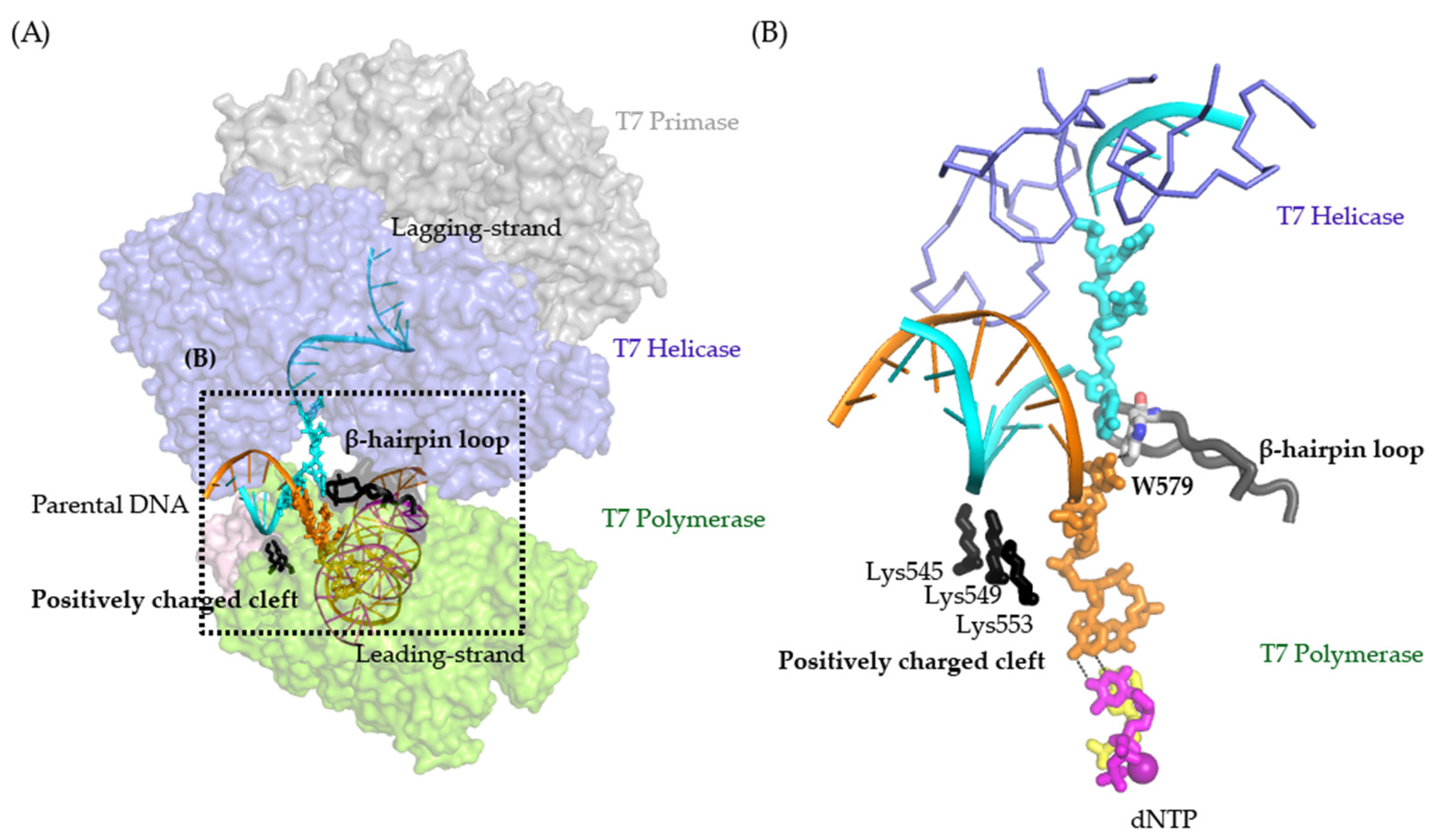
2.3. Synergistic Hel-Pol Binding to Fork DNA
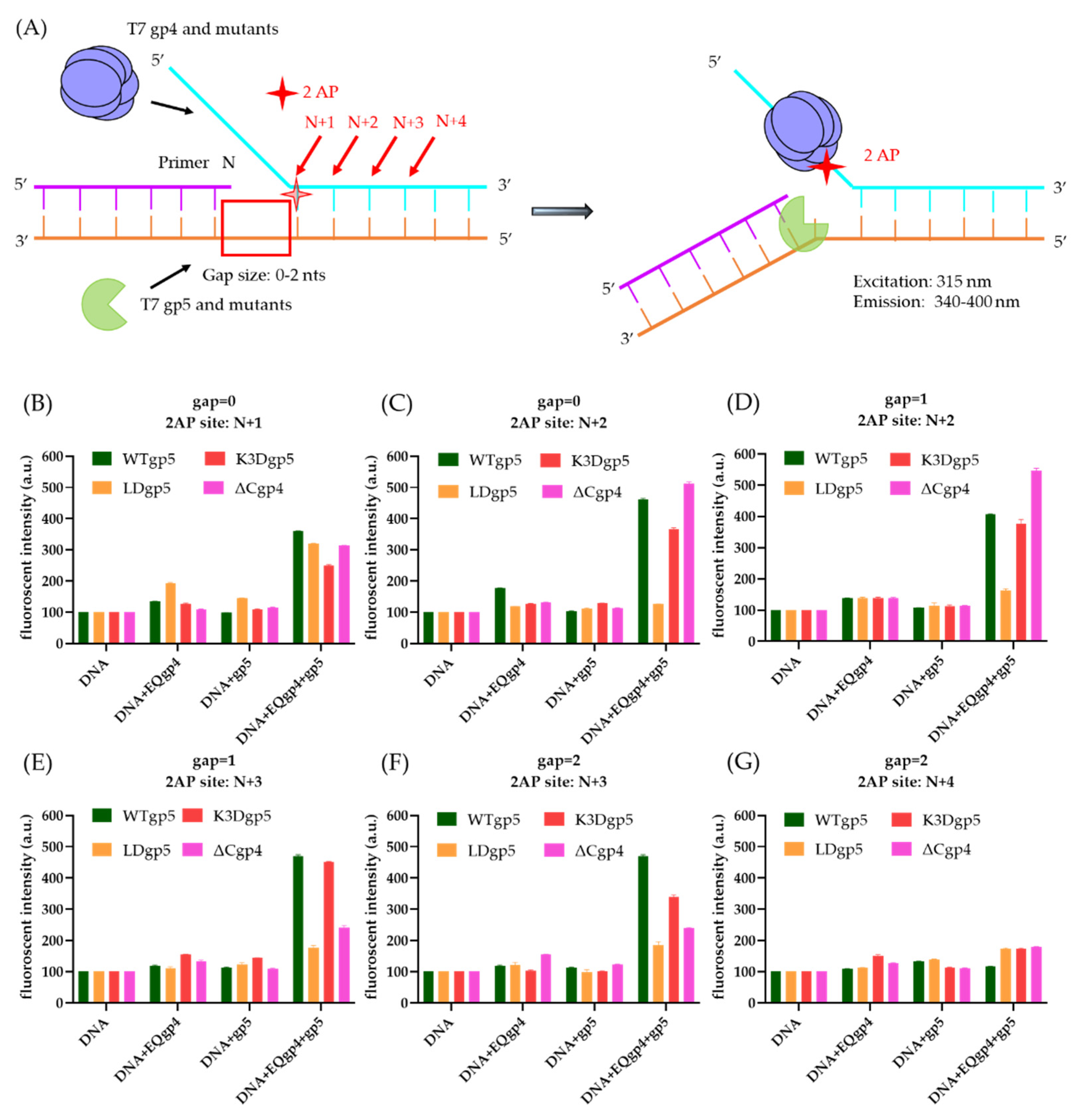
2.4. Cooperative gp4-gp5 Binding Induces Fork Unwinding
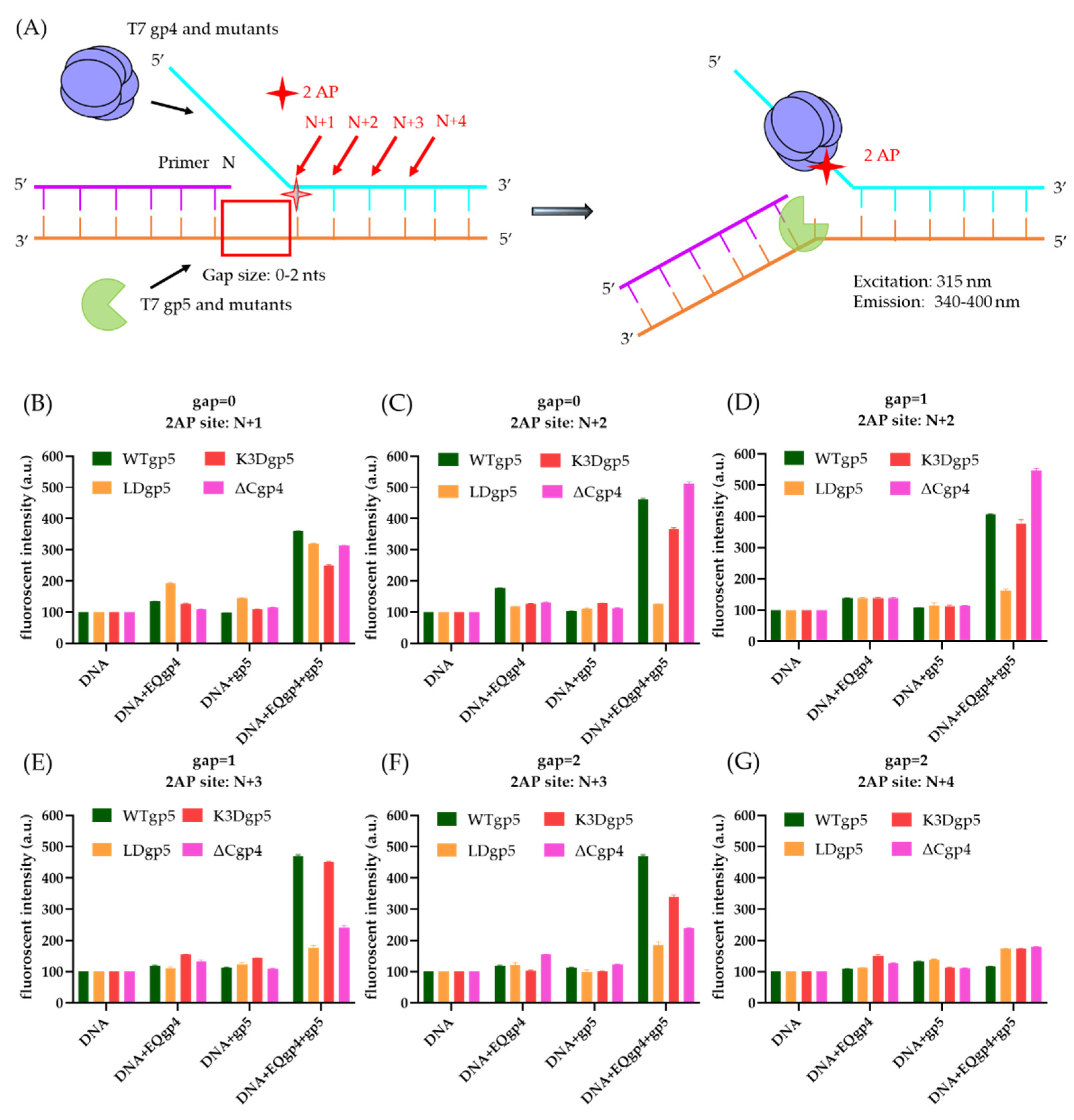
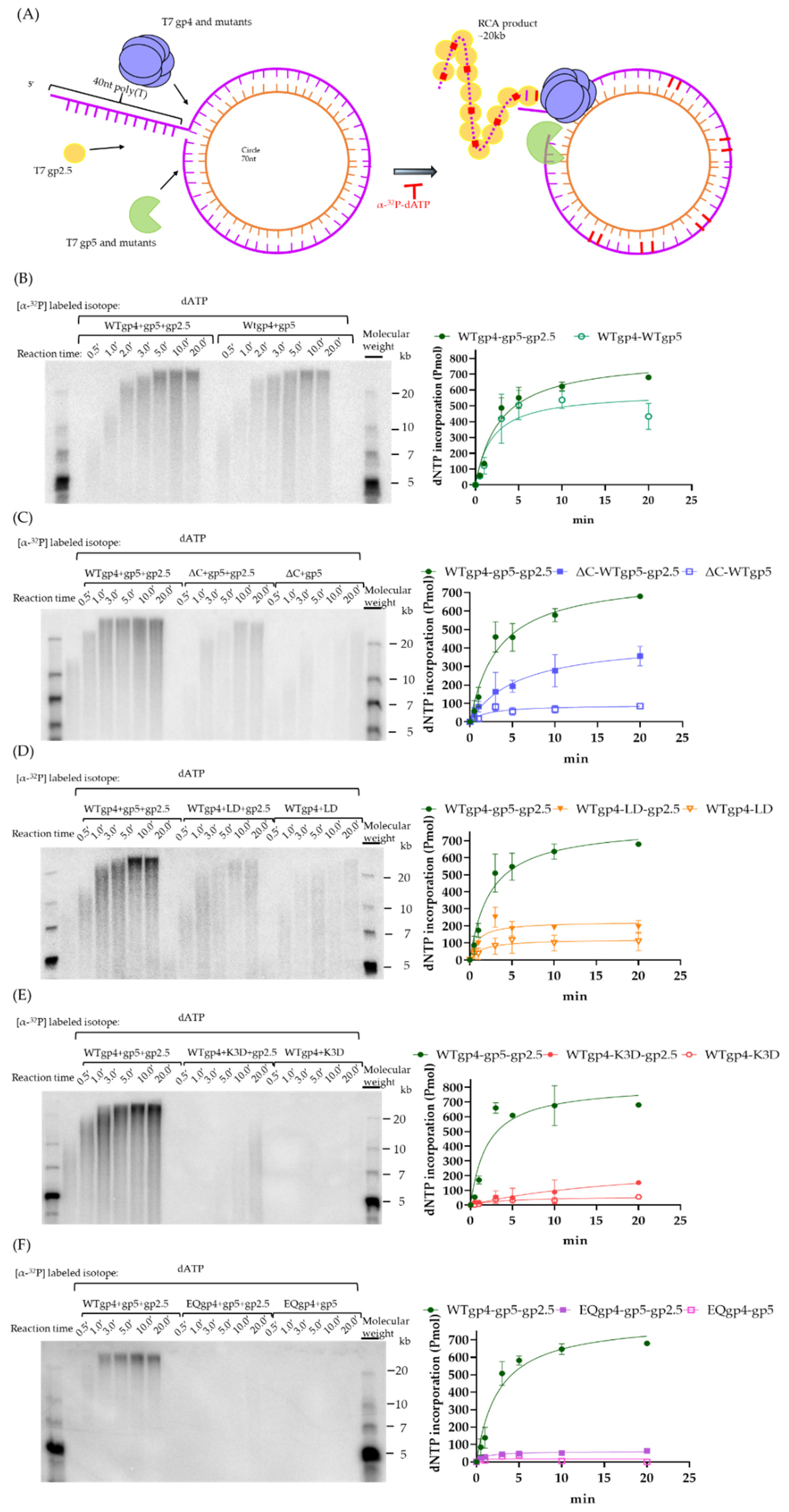
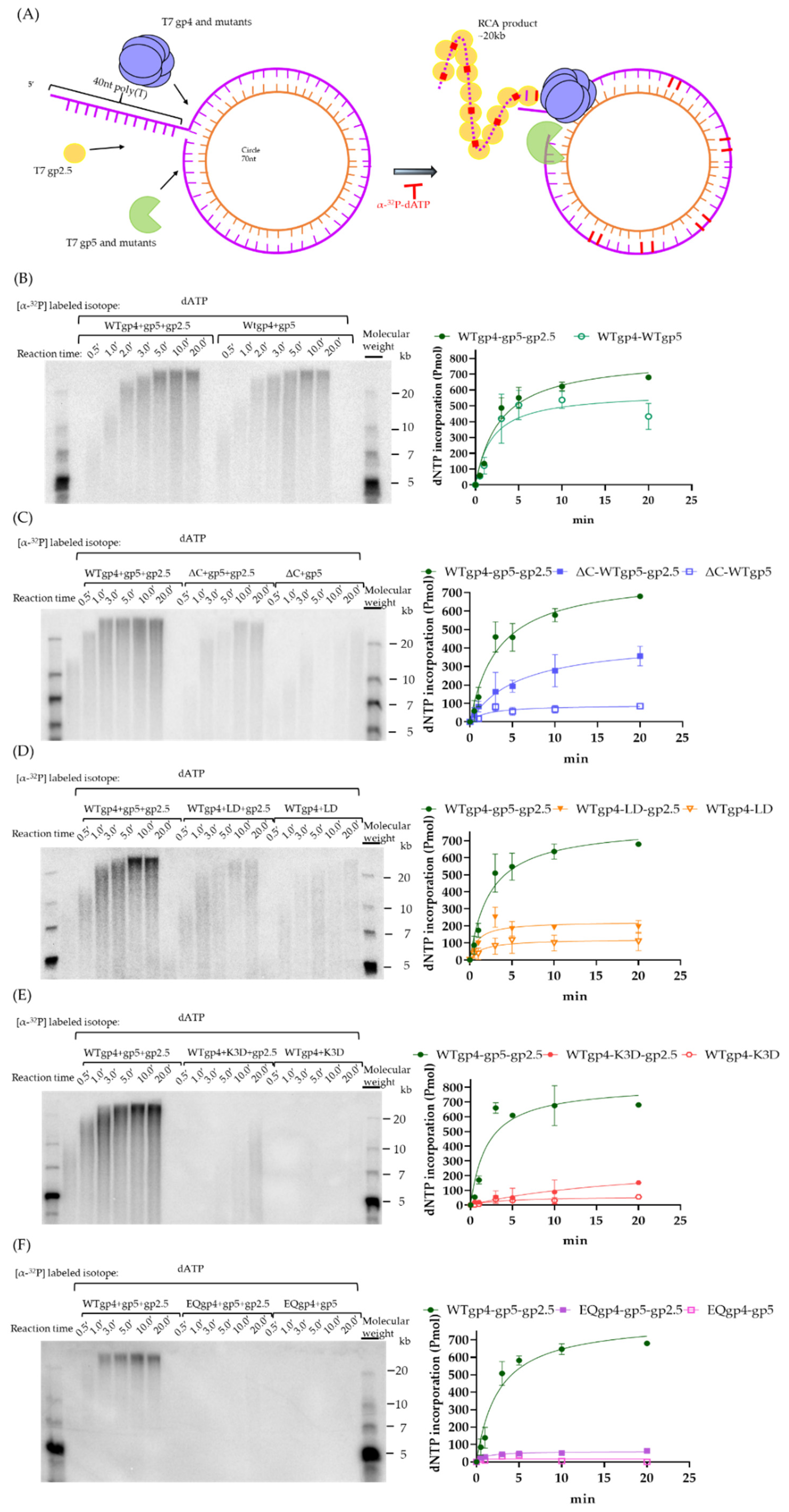
2.5. Coupled DNA Synthesis by T7 Replisome
3. Discussion
4. Materials and Methods
4.1. Clone, Expression and Purification of gp5-trx Complex
4.2. Clone, Expression, and Purification of gp4 Complex
4.3. DNA Polymerase Assays
4.4. Electrophoretic Mobility Shift Assays
4.5. 2-Aminopurine (2-AP) Assay
4.6. Rolling Circle Amplification
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a010108. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Richardson, C.C. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009, 78, 205–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkovic, S.J.; Valentine, A.M.; Salinas, F. Replisome-mediated DNA replication. Annu. Rev. Biochem. 2001, 70, 181–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- O’Donnell, M.E.; Li, H. The ring-shaped hexameric helicases that function at DNA replication forks. Nat. Struct. Mol. Biol. 2018, 25, 122–130. [Google Scholar] [CrossRef]
- Maffeo, C.; Aksimentiev, A. Molecular mechanism of DNA association with single-stranded DNA binding protein. Nucleic Acids Res. 2017, 45, 12125–12139. [Google Scholar] [CrossRef] [Green Version]
- Manosas, M.; Spiering, M.M.; Ding, F.; Croquette, V.; Benkovic, S.J. Collaborative coupling between polymerase and helicase for leading-strand synthesis. Nucleic Acids Res. 2012, 40, 6187–6198. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, D.; Pandey, M.; Patel, S.S. Cooperative base pair melting by helicase and polymerase positioned one nucleotide from each other. Elife 2015, 4, e06562. [Google Scholar] [CrossRef]
- Hamdan, S.M.; Johnson, D.E.; Tanner, N.A.; Lee, J.B.; Qimron, U.; Tabor, S.; van Oijen, A.M.; Richardson, C.C. Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 2007, 27, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.E.; Marians, K.J.; Kowalczykowski, S.C. Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell 2017, 169, 1201–1213. [Google Scholar] [CrossRef] [Green Version]
- Langston, L.D.; Zhang, D.; Yurieva, O.; Georgescu, R.E.; Finkelstein, J.; Yao, N.Y.; Indiani, C.; O’Donnell, M.E. CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc. Natl. Acad. Sci. USA 2014, 111, 15390–15395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Yang, W.; Seidman, M.M.; Rupp, W.D.; Gao, Y. Replisome structure suggests mechanism for continuous fork progression and post-replication repair. DNA Repair 2019, 81, 102658. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, W. Different mechanisms for translocation by monomeric and hexameric helicases. Curr. Opin. Struct. Biol. 2020, 61, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Enemark, E.J.; Joshua-Tor, L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 2006, 442, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Crampton, D.J.; Mukherjee, S.; Richardson, C.C. DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell 2006, 21, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.J.; Waksman, G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005, 71, 401–440. [Google Scholar]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Steitz, T.A. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar]
- Makarova, K.S.; Krupovic, M.; Koonin, E.V. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front. Microbiol. 2014, 5, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, C.M.; Potapova, O.; Delucia, A.M.; Huang, X.; Basu, V.P.; Grindley, N.D. Fingers-closing and other rapid conformational changes in DNA polymerase I (Klenow fragment) and their role in nucleotide selectivity. Biochemistry 2008, 47, 6103–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, G.W.; Hohlbein, J.; Craggs, T.; Aigrain, L.; Kapanidis, A.N. Real-time single-molecule studies of the motions of DNA polymerase fingers illuminate DNA synthesis mechanisms. Nucleic Acids Res. 2015, 43, 5998–6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Cui, Y.; Fox, T.; Lin, S.; Wang, H.; de Val, N.; Zhou, Z.H.; Yang, W. Structures and operating principles of the replisome. Science 2019, 363, eaav7003. [Google Scholar] [CrossRef]
- Yuan, Z.; Georgescu, R.; Schauer, G.D.; O’Donnell, M.E.; Li, H. Structure of the polymerase epsilon holoenzyme and atomic model of the leading strand replisome. Nat. Commun. 2020, 11, 3156. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, Y.; Georgescu, R.E.; Yuan, Z.; Chait, B.T.; Li, H.; O’Donnell, M.E. The architecture of a eukaryotic replisome. Nat. Struct. Mol. Biol. 2015, 22, 976–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manosas, M.; Spiering, M.M.; Ding, F.; Bensimon, D.; Allemand, J.F.; Benkovic, S.J.; Croquette, V. Mechanism of strand displacement synthesis by DNA replicative polymerases. Nucleic Acids Res. 2012, 40, 6174–6186. [Google Scholar] [CrossRef]
- Lo, C.Y.; Gao, Y. DNA Helicase-Polymerase Coupling in Bacteriophage DNA Replication. Viruses 2021, 13, 1739. [Google Scholar] [CrossRef]
- Zhang, H.; Lee, S.J.; Kulczyk, A.W.; Zhu, B.; Richardson, C.C. Heterohexamer of 56- and 63-kDa Gene 4 Helicase-Primase of Bacteriophage T7 in DNA Replication. J. Biol. Chem. 2012, 287, 34273–34287. [Google Scholar] [CrossRef] [Green Version]
- Doublie, S.; Tabor, S.; Long, A.M.; Richardson, C.C.; Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 1998, 391, 251–258. [Google Scholar] [CrossRef]
- Ghosh, S.; Hamdan, S.M.; Cook, T.E.; Richardson, C.C. Interactions of Escherichia coli thioredoxin, the processivity factor, with bacteriophage T7 DNA polymerase and helicase. J. Biol. Chem. 2008, 283, 32077–32084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, H.E.; Russel, M.; Model, P.; Richardson, C.C. Interaction of mutant thioredoxins of Escherichia coli with the gene 5 protein of phage T7. The redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J. Biol. Chem. 1986, 261, 15006–15012. [Google Scholar] [CrossRef]
- Hernandez, A.J.; Richardson, C.C. Gp2.5, the multifunctional bacteriophage T7 single-stranded DNA binding protein. Semin. Cell Dev. Biol. 2019, 86, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Quintero, V.; Peralta-Castro, A.; Benitez Cardoza, C.G.; Ellenberger, T.; Brieba, L.G. Structure of an open conformation of T7 DNA polymerase reveals novel structural features regulating primer-template stabilization at the polymerization active site. Biochem. J. 2021, 478, 2665–2679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lee, S.J.; Zhu, B.; Tran, N.Q.; Tabor, S.; Richardson, C.C. Helicase-DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 9372–9377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapathy, A.K.; Richardson, C.C. The glutamate switch of bacteriophage T7 DNA helicase: Role in coupling nucleotide triphosphate (NTP) and DNA binding to NTP hydrolysis. J. Biol. Chem. 2011, 286, 23113–23120. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Chastain, P.D., 2nd; Kusakabe, T.; Griffith, J.D.; Richardson, C.C. Coordinated leading and lagging strand DNA synthesis on a minicircular template. Mol. Cell 1998, 1, 1001–1010. [Google Scholar] [CrossRef]
- Spenkelink, L.M.; Spinks, R.R.; Jergic, S.; Lewis, J.S.; Dixon, N.E.; van Oijen, A.M. The E. coli helicase does not use ATP during replication. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yang, W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell 2006, 127, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Velankar, S.S.; Soultanas, P.; Dillingham, M.S.; Subramanya, H.S.; Wigley, D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 1999, 97, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, Y.; Lee, S.J.; Wei, Z.; Cao, J.; Richardson, C.C. Binding Affinities among DNA Helicase-Primase, DNA Polymerase, and Replication Intermediates in the Replisome of Bacteriophage T7. J. Biol. Chem. 2016, 291, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, I.; Lazaro, J.M.; Salas, M.; de Vega, M. Involvement of the TPR2 subdomain movement in the activities of phi29 DNA polymerase. Nucleic Acids Res. 2009, 37, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamtekar, S.; Berman, A.J.; Wang, J.; Lazaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage phi29. Mol. Cell 2004, 16, 609–618. [Google Scholar] [CrossRef]
- Morin, J.A.; Cao, F.J.; Lazaro, J.M.; Arias-Gonzalez, J.R.; Valpuesta, J.M.; Carrascosa, J.L.; Salas, M.; Ibarra, B. Active DNA unwinding dynamics during processive DNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 8115–8120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baretic, D.; Jenkyn-Bedford, M.; Aria, V.; Cannone, G.; Skehel, M.; Yeeles, J.T.P. Cryo-EM Structure of the Fork Protection Complex Bound to CMG at a Replication Fork. Mol. Cell 2020, 78, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Lionnet, T.; Spiering, M.M.; Benkovic, S.J.; Bensimon, D.; Croquette, V. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 19790–19795. [Google Scholar] [CrossRef] [Green Version]
- Manosas, M.; Xi, X.G.; Bensimon, D.; Croquette, V. Active and passive mechanisms of helicases. Nucleic Acids Res. 2010, 38, 5518–5526. [Google Scholar] [CrossRef] [Green Version]
- Syed, S.; Pandey, M.; Patel, S.S.; Ha, T. Single-molecule fluorescence reveals the unwinding stepping mechanism of replicative helicase. Cell Rep. 2014, 6, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Richardson, C.C. Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 2011, 15, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Satapathy, A.K.; Kulczyk, A.W.; Ghosh, S.; van Oijen, A.M.; Richardson, C.C. Coupling dTTP hydrolysis with DNA unwinding by the DNA helicase of bacteriophage T7. J. Biol. Chem. 2011, 286, 34468–34478. [Google Scholar] [CrossRef] [Green Version]
- Stano, N.M.; Jeong, Y.J.; Donmez, I.; Tummalapalli, P.; Levin, M.K.; Patel, S.S. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 2005, 435, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Kropp, H.M.; Durr, S.L.; Peter, C.; Diederichs, K.; Marx, A. Snapshots of a modified nucleotide moving through the confines of a DNA polymerase. Proc. Natl. Acad. Sci. USA 2018, 115, 9992–9997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Pandey, M.; Inman, J.T.; Yang, Y.; Kashlev, M.; Patel, S.S.; Wang, M.D. T7 replisome directly overcomes DNA damage. Nat. Commun. 2015, 6, 10260. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WTgp5 | WAgp5 | LDgp5 | K3Dgp5 | |
|---|---|---|---|---|
Linear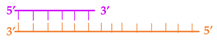 | 53 ± 5 nM | 48 ± 3 nM | 99 ± 3 nM | 170 ± 6 nM |
2gap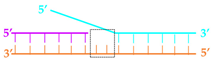 | 36 ± 5 nM | 66 ± 4 nM | 127 ± 11 nM | 112 ± 18 nM |
1gap | 63 ± 4 nM | 84 ± 16 nM | 139 ± 23 nM | 163 ± 2 nM |
0gap | 110 ± 13 nM | 81 ± 5 nM | 136 ± 9 nM | 214 ± 9 nM |
| DNA | DNA-WTgp5 | DNA-LDgp5 | DNA-K3Dgp5 | |
|---|---|---|---|---|
| EQgp4 | 24 ± 3 nM | 8 ± 1 nM | 25 ± 5 nM | 6 ± 1 nM |
| Control EQgp4ΔC | 30 ± 1 nM | 26 ± 1 nM | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo, C.-Y.; Gao, Y. DNA Polymerase-Parental DNA Interaction Is Essential for Helicase-Polymerase Coupling during Bacteriophage T7 DNA Replication. Int. J. Mol. Sci. 2022, 23, 1342. https://doi.org/10.3390/ijms23031342
Lo C-Y, Gao Y. DNA Polymerase-Parental DNA Interaction Is Essential for Helicase-Polymerase Coupling during Bacteriophage T7 DNA Replication. International Journal of Molecular Sciences. 2022; 23(3):1342. https://doi.org/10.3390/ijms23031342
Chicago/Turabian StyleLo, Chen-Yu, and Yang Gao. 2022. "DNA Polymerase-Parental DNA Interaction Is Essential for Helicase-Polymerase Coupling during Bacteriophage T7 DNA Replication" International Journal of Molecular Sciences 23, no. 3: 1342. https://doi.org/10.3390/ijms23031342
APA StyleLo, C.-Y., & Gao, Y. (2022). DNA Polymerase-Parental DNA Interaction Is Essential for Helicase-Polymerase Coupling during Bacteriophage T7 DNA Replication. International Journal of Molecular Sciences, 23(3), 1342. https://doi.org/10.3390/ijms23031342