
Pericytes of Stria Vascularis Are Targets of Cisplatin-Induced Ototoxicity: New Insights into the Molecular Mechanisms Involved in Blood-Labyrinth Barrier Breakdown
 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
- (1)
- to evaluate the effect of cisplatin on bovine cochlear pericytes (BCPs), which anatomically participate in BLB formation, both in the absence and in the presence of dexamethasone;
- (2)
- to suggest further studies on a new molecule, PDGF-β, for a future therapy based on the restoration/maintenance of the vitality of the pericytes that make up the SV.
2. Results
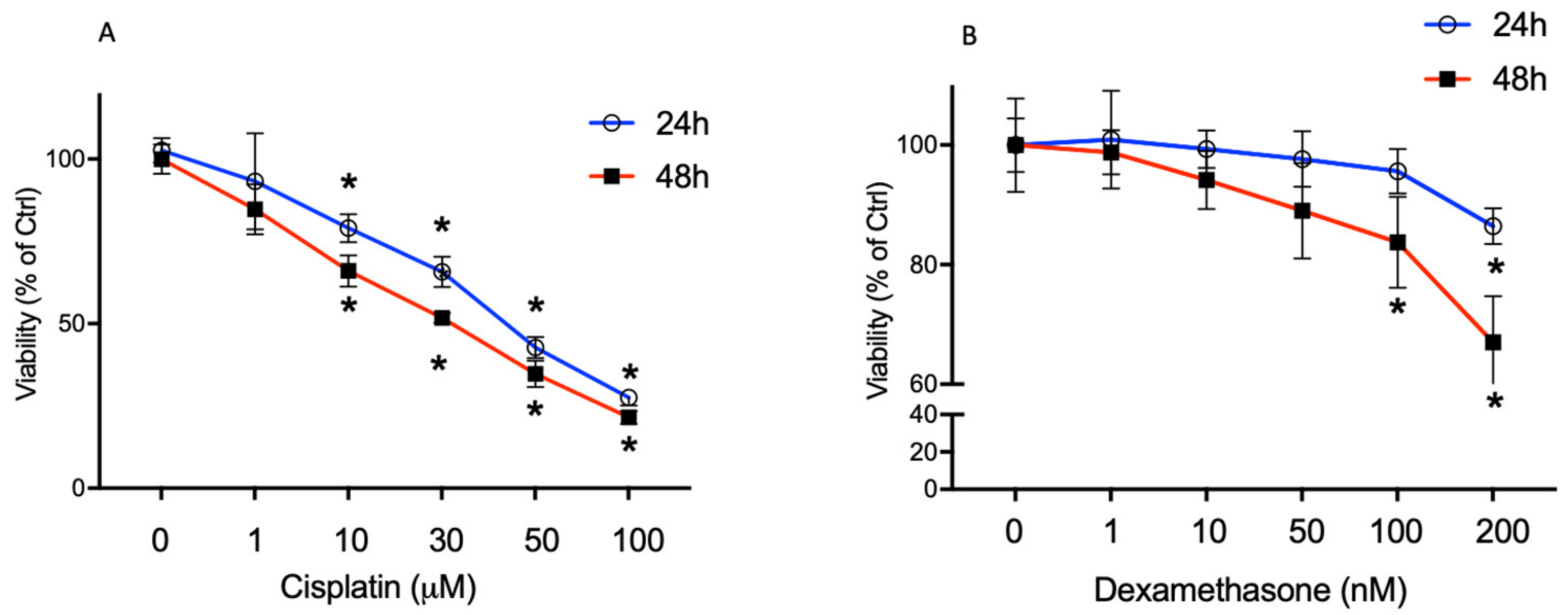
2.1. High Cisplatin Concentrations Affected Cell Viability
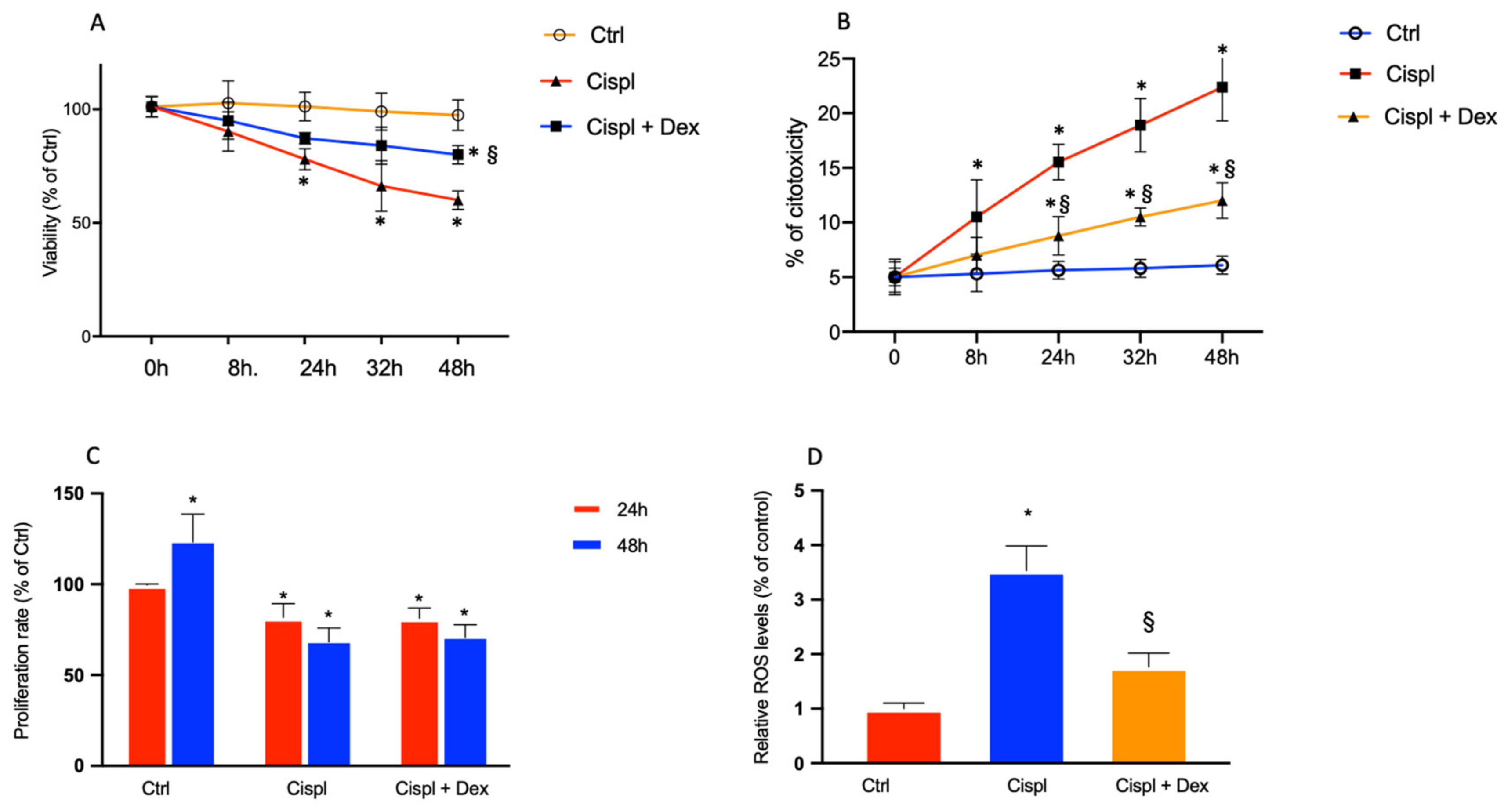
2.2. Dexamethasone Elicits a Protective Effect on Cisplatin-Treated BCPs
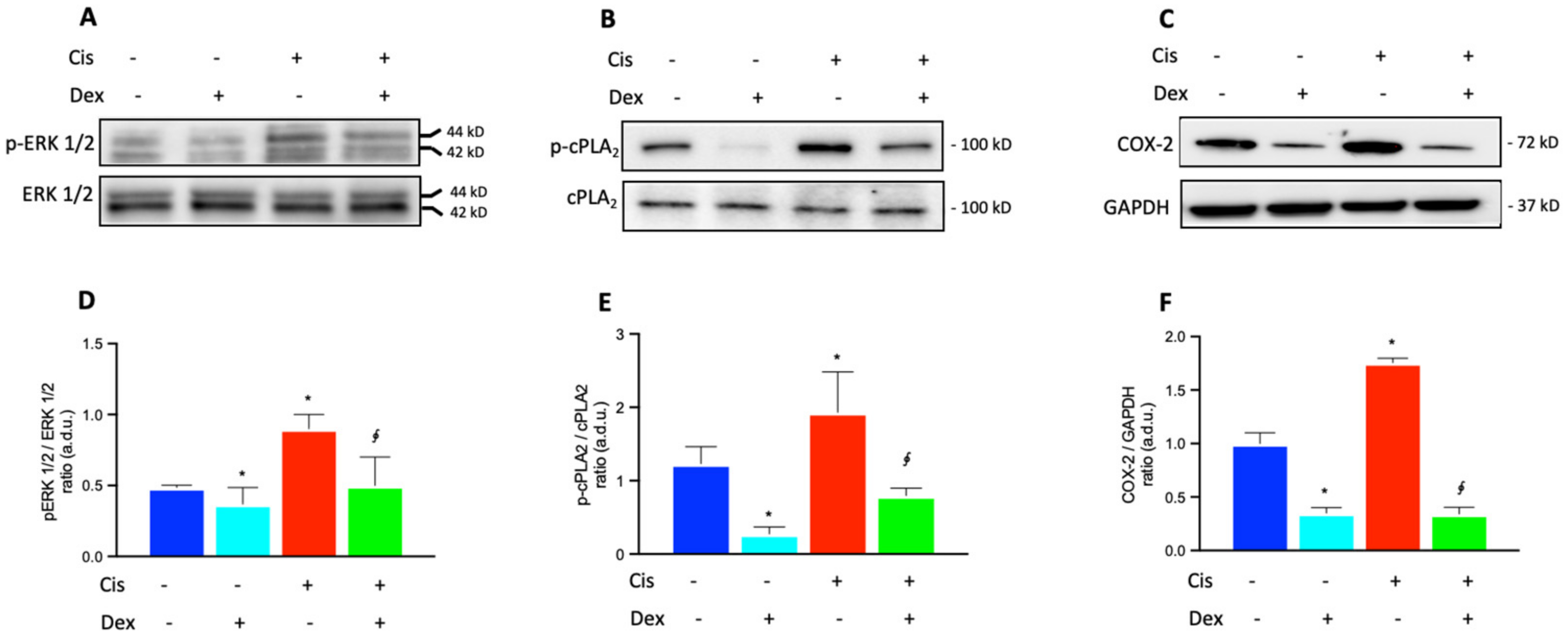
2.3. Dexamethasone Up-Regulates the ERK1/2/cPLA2/COX-2 Axis in BCPs Treated with Cisplatin
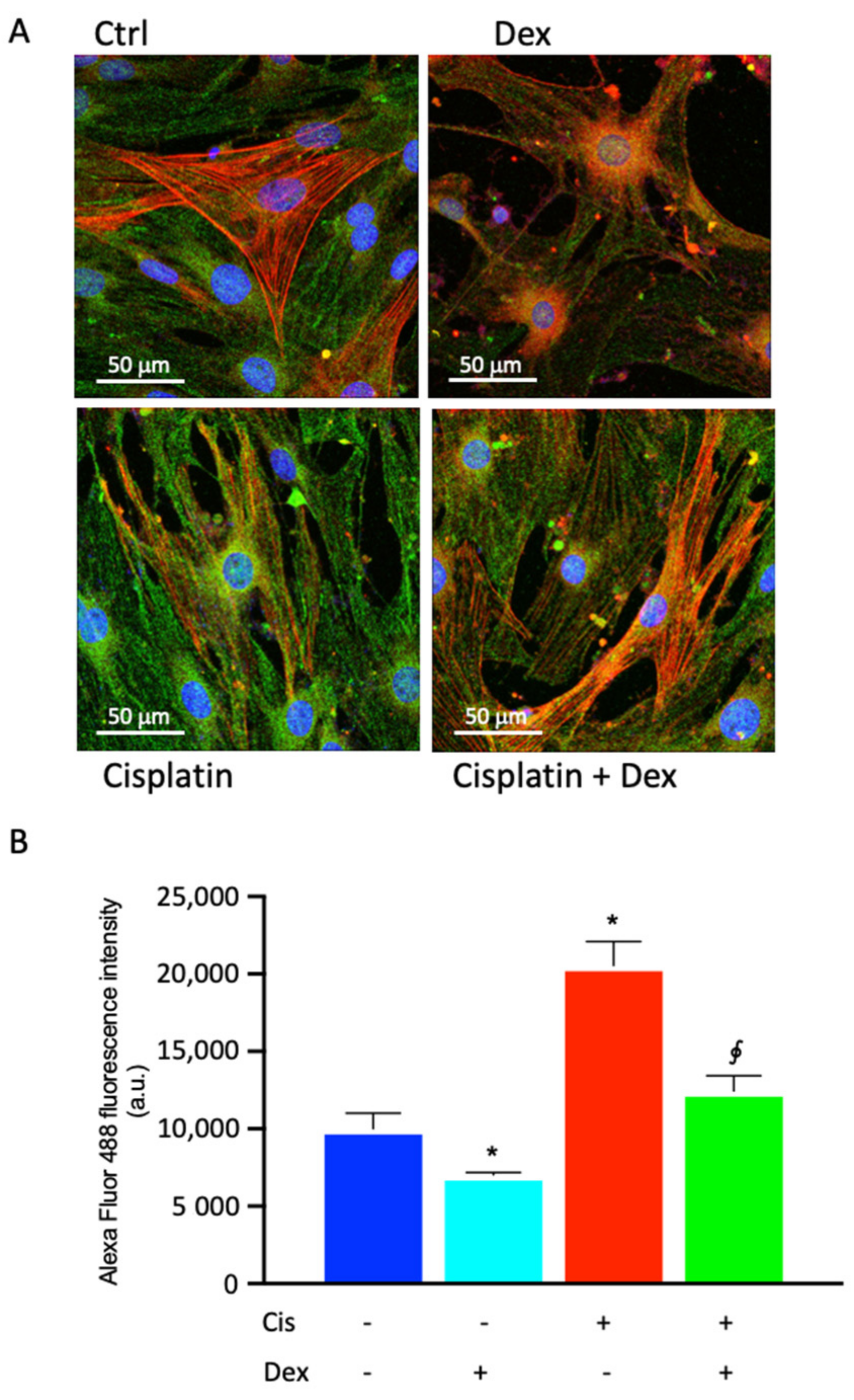
2.4. Dexamethasone Affected COX-2 Fluorescence in BCPs Treated with Cisplatin
2.5. Prostaglandin Production and VEGFA Release in BCPs Stimulated and Not Stimulated by Cisplatin in the Absence or in the Presence of Dexamethasone
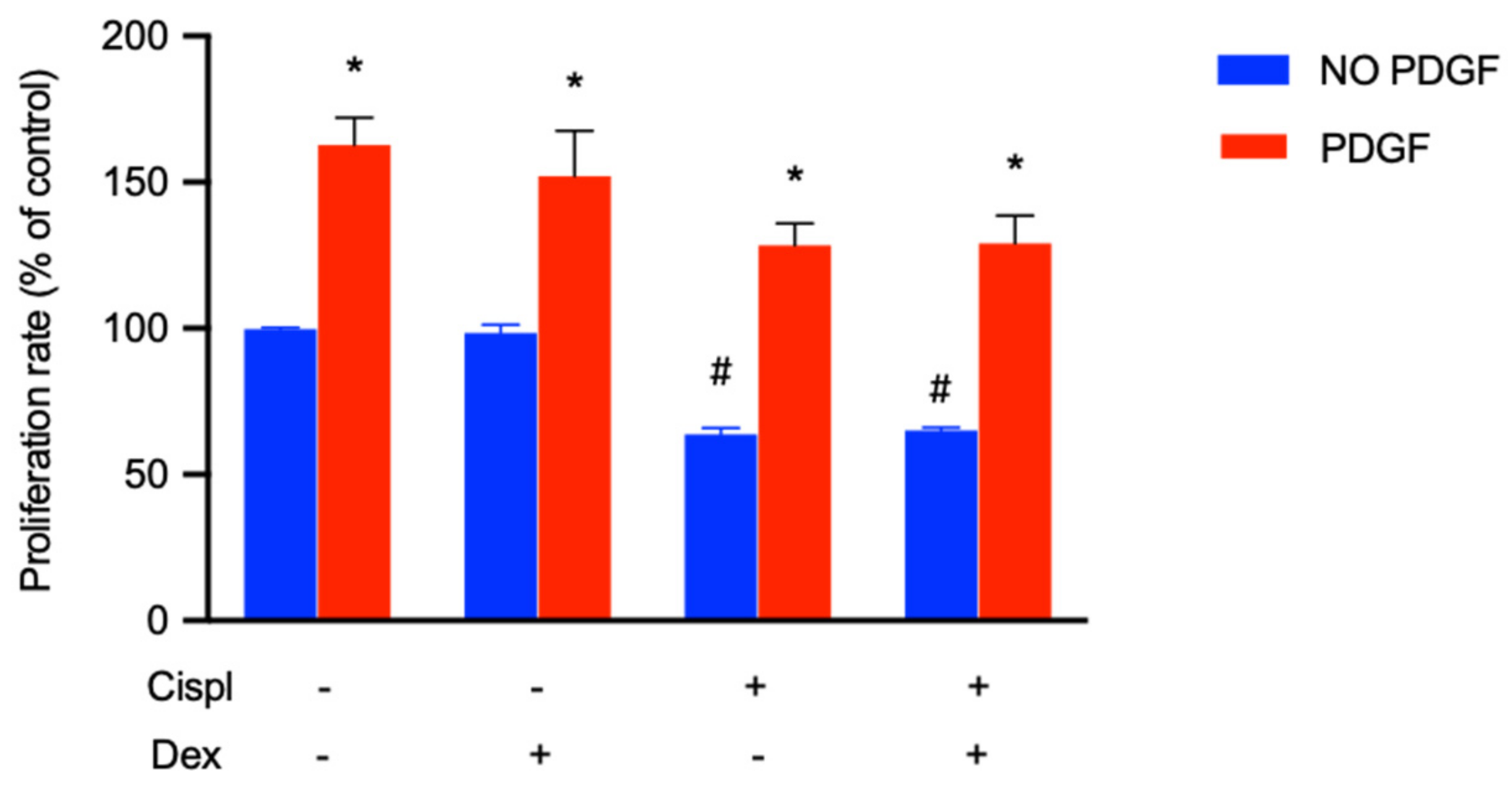
2.6. Protective Effect of PDGF-BB on BCPs Treated with Cisplatin
3. Discussion
- (i)
- cisplatin induced ROS formation also in BCPs, and therefore the production of ROS, which contributes to making cisplatin very efficient against cancer cells, also has negative effects on non-cancer cells; this is undoubtedly one of the side effects of this chemotherapy;
- (ii)
- Dex, used to counteract cisplatin-induced hearing loss by reducing free radical levels, as we have shown here, could represent one mechanism that reduces the anticancer effects of cisplatin. Indeed, Dex has been shown to reduce the anticancer effects of a drug [27], and can create resistance to chemotherapy through different mechanisms, for example by increasing the adhesion of human ovarian cancer cell lines to the extracellular matrix [52] or by up-regulating Krüppel-like factor 5 in triple-negative breast cancer [53]. The reduction in free radical production induced by Dex could be another mechanism that reduces the antitumor activity of cisplatin.
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Experimental Protocol
4.3. Cell Viability
4.4. Cell Proliferation Assay
4.5. Western Blotting
4.6. Confocal Microscopy
4.7. ROS Measurements
4.8. Prostaglandin E2 Production and VEGF Release
4.9. Statistical Analysis
5. Conclusions
- (1)
- BCPs are a target of cisplatin damage: the reduction of their viability could cause a decrease in PGE2 production, with severe implications for microcapillary permeability;
- (2)
- one of the mechanisms by which cisplatin performs anticancer activity is the production of ROS, which leads to the death of cancer cells. Dex, in the presence of cisplatin, reduced ROS production and this mechanism could interfere with the antitumor activity of cisplatin;
- (3)
- cisplatin triggered an inflammatory process in BCPs by activating p-ERK 1/2, p-cPLA2 and COX-2 and inducing an increase in the release of PGE2. Dex reduced PGE2 production and therefore reduced the modulation of SV permeability;
- (4)
- the treatment of BCPs with PDGF-BB induced a recovery of their proliferation in the presence of cisplatin. The therapeutic use of PDGF-BB could allow for the replenishment of PGE2 and the maintenance of the BLB structure.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, W.; Zong, S.; Du, P.; Zhou, P.; Li, H.; Wang, E.; Xiao, H. Role of the Stria Vascularis in the Pathogenesis of Sensorineural Hearing Loss: A Narrative Review. Front. Neurosci. 2021, 15, 774585. [Google Scholar] [CrossRef] [PubMed]
- Keithley, E.M. Pathology and Mechanisms of Cochlear Aging. J. Neurosci. Res. 2020, 98, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Hou, Z.; Neng, L.; Cai, J.; Zhang, Y.; Shi, X. Suppression of Connexin 43 Leads to Strial Vascular Hyper-Permeability, Decrease in Endocochlear Potential, and Mild Hearing Loss. Front. Physiol. 2020, 11, 974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ota, T.; Yoshida, T.; Ino, D.; Sato, M.P.; Doi, K.; Horii, A.; Nin, F.; Hibino, H. Electrochemical Properties of the Non-excitable Tissue Stria Vascularis of the Mammalian Cochlea Are Sensitive to Sounds. J. Physiol. 2021, 599, 4497–4516. [Google Scholar] [CrossRef] [PubMed]
- Shi, X. Pathophysiology of the Cochlear Intrastrial Fluid-Blood Barrier (Review). Hear. Res. 2016, 338, 52–63. [Google Scholar] [CrossRef]
- Neng, L.; Zhang, F.; Kachelmeier, A.; Shi, X. Endothelial Cell, Pericyte, and Perivascular Resident Macrophage-Type Melanocyte Interactions Regulate Cochlear Intrastrial Fluid–Blood Barrier Permeability. J. Assoc. Res. Otolaryngol. 2013, 14, 175–185. [Google Scholar] [CrossRef]
- Cohen-Salmon, M.; Regnault, B.; Cayet, N.; Caille, D.; Demuth, K.; Hardelin, J.-P.; Janel, N.; Meda, P.; Petit, C. Connexin30 Deficiency Causes Instrastrial Fluid-Blood Barrier Disruption within the Cochlear Stria Vascularis. Proc. Natl. Acad. Sci. USA 2007, 104, 6229–6234. [Google Scholar] [CrossRef]
- Shi, X.; Han, W.; Yamamoto, H.; Tang, W.; Lin, X.; Xiu, R.; Trune, D.R.; Nuttall, A.L. The Cochlear Pericytes. Microcirculation 2008, 15, 515–529. [Google Scholar] [CrossRef]
- Canis, M.; Bertlich, M. Cochlear Capillary Pericytes. In Pericyte Biology in Different Organs; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1122, pp. 115–123. ISBN 978-3-030-11092-5. [Google Scholar]
- Neng, L.; Zhang, J.; Yang, J.; Zhang, F.; Lopez, I.A.; Dong, M.; Shi, X. Structural Changes in Thestrial Blood-Labyrinth Barrier of Aged C57BL/6 Mice. Cell Tissue Res. 2015, 361, 685–696. [Google Scholar] [CrossRef]
- Shi, X. Cochlear Pericyte Responses to Acoustic Trauma and the Involvement of Hypoxia-Inducible Factor-1alpha and Vascular Endothelial Growth Factor. Am. J. Pathol. 2009, 174, 1692–1704. [Google Scholar] [CrossRef]
- Darland, D.C.; Massingham, L.J.; Smith, S.R.; Piek, E.; Saint-Geniez, M.; D’Amore, P.A. Pericyte Production of Cell-Associated VEGF Is Differentiation-Dependent and Is Associated with Endothelial Survival. Dev. Biol. 2003, 264, 275–288. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, Z.; Wang, X.; Jiang, H.; Neng, L.; Zhang, Y.; Yu, Q.; Burwood, G.; Song, J.; Auer, M.; et al. VEGFA165 Gene Therapy Ameliorates Blood-Labyrinth Barrier Breakdown and Hearing Loss. JCI Insight 2021, 6, e143285. [Google Scholar] [CrossRef]
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived Growth Factor-BB Promotes Proliferation and Migration of Retinal Microvascular Pericytes by Up-regulating the Expression of C-X-C Chemokine Receptor Types 4. Exp. Ther. Med. 2019, 18, 4022–4030. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Babic, S.; De Gooyer, T.; Stitt, A.W.; Jaworski, K.; Ong, L.G.T.; Kelly, D.J.; Gilbert, R.E. Inhibition of Platelet-Derived Growth Factor Promotes Pericyte Loss and Angiogenesis in Ischemic Retinopathy. Am. J. Pathol. 2004, 164, 1263–1273. [Google Scholar] [CrossRef]
- Thulasiram, M.R.; Ogier, J.M.; Dabdoub, A. Hearing Function, Degeneration, and Disease: Spotlight on the Stria Vascularis. Front. Cell Dev. Biol. 2022, 10, 841708. [Google Scholar] [CrossRef]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef]
- Dulon, D.; Aran, J.M.; Zajic, G.; Schacht, J. Comparative Uptake of Gentamicin, Netilmicin, and Amikacin in the Guinea Pig Cochlea and Vestibule. Antimicrob. Agents Chemother. 1986, 30, 96–100. [Google Scholar] [CrossRef]
- Caporarello, N.; D’Angeli, F.; Cambria, M.T.; Candido, S.; Giallongo, C.; Salmeri, M.; Lombardo, C.; Longo, A.; Giurdanella, G.; Anfuso, C.D.; et al. Pericytes in Microvessels: From “Mural” Function to Brain and Retina Regeneration. Int. J. Mol. Sci. 2019, 20, 6351. [Google Scholar] [CrossRef]
- Nyberg, S.; Abbott, N.J.; Shi, X.; Steyger, P.S.; Dabdoub, A. Delivery of Therapeutics to the Inner Ear: The Challenge of the Blood-Labyrinth Barrier. Sci. Transl. Med. 2019, 11, eaao0935. [Google Scholar] [CrossRef]
- Zhang, N.; Cai, J.; Xu, L.; Wang, H.; Liu, W. Cisplatin-Induced Stria Vascularis Damage Is Associated with Inflammation and Fibrosis. Neural Plast. 2020, 2020, 8851525. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Yao, X.; Li, W.; Du, H.; Tang, M.; Xiong, W.; Chai, R.; Xu, Z. Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front. Mol. Neurosci. 2017, 10, 401. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Mechanisms of Cisplatin-Induced Ototoxicity and Otoprotection. Front. Cell. Neurosci. 2017, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Lieu, J.E.C.; Kenna, M.; Anne, S.; Davidson, L. Hearing Loss in Children: A Review. JAMA 2020, 324, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P.; Dhukhwa, A.; Mukherjea, D.; Ramkumar, V. Local Drug Delivery for Prevention of Hearing Loss. Front. Cell. Neurosci. 2019, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Taukulis, I.A.; Olszewski, R.T.; Korrapati, S.; Fernandez, K.A.; Boger, E.T.; Fitzgerald, T.S.; Morell, R.J.; Cunningham, L.L.; Hoa, M. Single-Cell RNA-Seq of Cisplatin-Treated Adult Stria Vascularis Identifies Cell Type-Specific Regulatory Networks and Novel Therapeutic Gene Targets. Front. Mol. Neurosci. 2021, 14, 718241. [Google Scholar] [CrossRef] [PubMed]
- Kros, C.J.; Steyger, P.S. Aminoglycoside- and Cisplatin-Induced Ototoxicity: Mechanisms and Otoprotective Strategies. Cold Spring Harb. Perspect. Med. 2019, 9, a033548. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Iavicoli, I.; Calabrese, V. Hormesis: Why It Is Important to Biogerontologists. Biogerontology 2012, 13, 215–235. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular Stress Responses, the Hormesis Paradigm, and Vitagenes: Novel Targets for Therapeutic Intervention in Neurodegenerative Disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef]
- Drake, J.; Sultana, R.; Aksenova, M.; Calabrese, V.; Butterfield, D.A. Elevation of Mitochondrial Glutathione by Gamma-Glutamylcysteine Ethyl Ester Protects Mitochondria against Peroxynitrite-Induced Oxidative Stress. J. Neurosci. Res. 2003, 74, 917–927. [Google Scholar] [CrossRef]
- Mirzaei, S.; Hushmandi, K.; Zabolian, A.; Saleki, H.; Torabi, S.M.R.; Ranjbar, A.; SeyedSaleh, S.; Sharifzadeh, S.O.; Khan, H.; Ashrafizadeh, M.; et al. cElucidating Role of Reactive Oxygen Species (ROS) in Cisplatin Chemotherapy: A Focus on Molecular Pathways and Possible Therapeutic Strategies. Molecules 2021, 26, 2382. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, W.; Sun, X.; Wang, Y.; Zhou, M. Kaempferol Ameliorates Cisplatin Induced Nephrotoxicity by Modulating Oxidative Stress, Inflammation and Apoptosis via ERK and NF-ΚB Pathways. AMB Express 2020, 10, 58. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Olivieri, M.; Fidilio, A.; Lupo, G.; Rusciano, D.; Pezzino, S.; Gagliano, C.; Drago, F.; Bucolo, C. Gabapentin Attenuates Ocular Inflammation: In Vitro and In Vivo Studies. Front. Pharmacol. 2017, 8, 173. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M.G. Nitric Oxide in the Central Nervous System: Neuroprotection versus Neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lupo, G.; Gennuso, F.; Conti, F.; Furno, D.L.; Mannino, G.; Anfuso, C.D.; Drago, F.; Salomone, S.; Bucolo, C. Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 7528. [Google Scholar] [CrossRef]
- Nicotra, A.; Lupo, G.; Giurdanella, G.; Anfuso, C.D.; Ragusa, N.; Tirolo, C.; Marchetti, B.; Alberghina, M. MAPKs Mediate the Activation of Cytosolic Phospholipase A2 by Amyloid Beta(25-35) Peptide in Bovine Retina Pericytes. Biochim. Biophys. Acta 2005, 1733, 172–186. [Google Scholar] [CrossRef]
- Yang, T. Microsomal Prostaglandin E Synthase-1 and Blood Pressure Regulation. Kidney Int. 2007, 72, 274–278. [Google Scholar] [CrossRef]
- Gao, Z.; Schwieger, J.; Matin-Mann, F.; Behrens, P.; Lenarz, T.; Scheper, V. Dexamethasone for Inner Ear Therapy: Biocompatibility and Bio-Efficacy of Different Dexamethasone Formulations In Vitro. Biomolecules 2021, 11, 1896. [Google Scholar] [CrossRef]
- Li, X.; Chen, W.-J.; Xu, J.; Yi, H.-J.; Ye, J.-Y. Clinical Analysis of Intratympanic Injection of Dexamethasone for Treating Sudden Deafness. Int. J. Gen. Med. 2021, 14, 2575–2579. [Google Scholar] [CrossRef]
- Wilk, M.; Hessler, R.; Mugridge, K.; Jolly, C.; Fehr, M.; Lenarz, T.; Scheper, V. Impedance Changes and Fibrous Tissue Growth after Cochlear Implantation Are Correlated and Can Be Reduced Using a Dexamethasone Eluting Electrode. PLoS ONE 2016, 11, e0147552. [Google Scholar] [CrossRef]
- Rutherford, R.B.; Ryan, M.E.; Kennedy, J.E.; Tucker, M.M.; Charette, M.F. Platelet-Derived Growth Factor and Dexamethasone Combined with a Collagen Matrix Induce Regeneration of the Periodontium in Monkeys. J. Clin. Periodontol. 1993, 20, 537–544. [Google Scholar] [CrossRef]
- Rutherford, R.B.; TrailSmith, M.D.; Ryan, M.E.; Charette, M.F. Synergistic Effects of Dexamethasone on Platelet-Derived Growth Factor Mitogenesis in Vitro. Arch. Oral Biol. 1992, 37, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Warshamana, G.S.; Martinez, S.; Lasky, J.A.; Corti, M.; Brody, A.R. Dexamethasone Activates Expression of the PDGF-α Receptor and Induces Lung Fibroblast Proliferation. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 274, L499–L507. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Neng, L.; Zhang, J.; Cai, J.; Wang, X.; Zhang, Y.; Lopez, I.A.; Shi, X. Acoustic Trauma Causes Cochlear Pericyte-to-Myofibroblast-Like Cell Transformation and Vascular Degeneration, and Transplantation of New Pericytes Prevents Vascular Atrophy. Am. J. Pathol. 2020, 190, 1943–1959. [Google Scholar] [CrossRef] [PubMed]
- Lyu, A.-R.; Kim, D.H.; Lee, S.H.; Shin, D.-S.; Shin, S.-A.; Park, Y.-H. Effects of Dexamethasone on Intracochlear Inflammation and Residual Hearing after Cochleostomy: A Comparison of Administration Routes. PLoS ONE 2018, 13, e0195230. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, G.; Lopez, I.A.; Ishiyama, P.; Vinters, H.V.; Ishiyama, A. The Blood Labyrinthine Barrier in the Human Normal and Meniere’s Disease Macula Utricle. Sci. Rep. 2017, 7, 253. [Google Scholar] [CrossRef]
- Barna, L.; Walter, F.R.; Harazin, A.; Bocsik, A.; Kincses, A.; Tubak, V.; Jósvay, K.; Zvara, Á.; Campos-Bedolla, P.; Deli, M.A. Simvastatin, Edaravone and Dexamethasone Protect against Kainate-Induced Brain Endothelial Cell Damage. Fluids Barriers CNS 2020, 17, 5. [Google Scholar] [CrossRef]
- Stone, N.L.; England, T.J.; O’Sullivan, S.E. A Novel Transwell Blood Brain Barrier Model Using Primary Human Cells. Front. Cell. Neurosci. 2019, 13, 230. [Google Scholar] [CrossRef]
- Choi, Y.-M.; Kim, H.-K.; Shim, W.; Anwar, M.A.; Kwon, J.-W.; Kwon, H.-K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef]
- Xue, D.-F.; Pan, S.-T.; Huang, G.; Qiu, J.-X. ROS Enhances the Cytotoxicity of Cisplatin by Inducing Apoptosis and Autophagy in Tongue Squamous Cell Carcinoma Cells. Int. J. Biochem. Cell Biol. 2020, 122, 105732. [Google Scholar] [CrossRef]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct Impact of Cisplatin on Mitochondria Induces ROS Production That Dictates Cell Fate of Ovarian Cancer Cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Wang, Y.; Fu, C.-C.; Diao, F.; Song, L.-N.; Li, Z.-B.; Yang, R.; Lu, J. Dexamethasone Enhances Cell Resistance to Chemotherapy by Increasing Adhesion to Extracellular Matrix in Human Ovarian Cancer Cells. Endocr. Relat. Cancer 2010, 17, 39–50. [Google Scholar] [CrossRef]
- Li, Z.; Dong, J.; Zou, T.; Du, C.; Li, S.; Chen, C.; Liu, R.; Wang, K. Dexamethasone Induces Docetaxel and Cisplatin Resistance Partially through Up-Regulating Krüppel-like Factor 5 in Triple-Negative Breast Cancer. Oncotarget 2017, 8, 11555–11565. [Google Scholar] [CrossRef]
- Nishimura, T.; Nario, K.; Hosoi, H. Effects of Intravenous Administration of Prostaglandin E(1) and Lipo-Prostaglandin E(1) on Cochlear Blood Flow in Guinea Pigs. Eur. Arch. Otorhinolaryngol. 2002, 259, 253–256. [Google Scholar] [CrossRef]
- Umemura, K.; Nakashima, M. Effect of Prostaglandin E1 on the Rat Inner Ear Microvascular Thrombosis. Gen. Pharmacol. 1997, 28, 221–224. [Google Scholar] [CrossRef]
- Ziegler, E.A.; Brieger, J.; Heinrich, U.R.; Mann, W.J. Immunohistochemical Localization of Cyclooxygenase Isoforms in the Organ of Corti and the Spiral Ganglion Cells of Guinea Pig Cochlea. ORL J. Otorhinolaryngol. Relat. Spec. 2004, 66, 297–301. [Google Scholar] [CrossRef]
- Heinrich, U.-R.; Brieger, J.; Selivanova, O.; Feltens, R.; Eimermacher, A.; Schäfer, D.; Mann, W.J. COX-2 Expression in the Guinea Pig Cochlea Is Partly Altered by Moderate Sound Exposure. Neurosci. Lett. 2006, 394, 121–126. [Google Scholar] [CrossRef]
- Denekamp, J.; Hobson, B. Endothelial-Cell Proliferation in Experimental Tumours. Br. J. Cancer 1982, 46, 711–720. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, H.; Zuo, L. Anti-Inflammatory Activities of a New VEGF Blocker, Conbercept. Immunopharmacol. Immunotoxicol. 2021, 43, 594–598. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.M.; Park, J.S.; Jo, S.A.; Kim, Y.-O.; Kim, C.-W.; Jo, I. Dexamethasone Coordinately Regulates Angiopoietin-1 and VEGF: A Mechanism of Glucocorticoid-Induced Stabilization of Blood–Brain Barrier. Biochem. Biophys. Res. Commun. 2008, 372, 243–248. [Google Scholar] [CrossRef]
- Cervantes, B.; Arana, L.; Murillo-Cuesta, S.; Bruno, M.; Alkorta, I.; Varela-Nieto, I. Solid Lipid Nanoparticles Loaded with Glucocorticoids Protect Auditory Cells from Cisplatin-Induced Ototoxicity. J. Clin. Med. 2019, 8, 1464. [Google Scholar] [CrossRef]
- Rivera, T.; Sanz, L.; Camarero, G.; Varela-Nieto, I. Drug Delivery to the Inner Ear: Strategies and Their Therapeutic Implications for Sensorineural Hearing Loss. Curr. Drug Deliv. 2012, 9, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wikström, A.C.; Bakke, O.; Okret, S.; Brönnegård, M.; Gustafsson, J.A. Intracellular Localization of the Glucocorticoid Receptor: Evidence for Cytoplasmic and Nuclear Localization. Endocrinology 1987, 120, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Albu, S.; Nagy, A.; Doros, C.; Marceanu, L.; Cozma, S.; Musat, G.; Trabalzini, F. Treatment of Meniere’s Disease with Intratympanic Dexamethazone plus High Dosage of Betahistine. Am. J. Otolaryngol. 2016, 37, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.P.C.; Stathopoulos, D.; O’Leary, S. Steroids for Idiopathic Sudden Sensorineural Hearing Loss. Cochrane Database Syst. Rev. 2013, CD003998. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.; Lovett, B.; Johns, J.D.; Gu, S.; Choi, D.; Trune, D.; Hoa, M. In Silico Single-Cell Analysis of Steroid-Responsive Gene Targets in the Mammalian Cochlea. Front. Neurol. 2021, 12, 818157. [Google Scholar] [CrossRef]
- Giurdanella, G.; Montalbano, G.; Gennuso, F.; Brancati, S.; Lo Furno, D.; Augello, A.; Bucolo, C.; Drago, F.; Salomone, S. Isolation, Cultivation, and Characterization of Primary Bovine Cochlear Pericytes: A New in Vitro Model of Stria Vascularis. J. Cell. Physiol. 2019, 234, 1978–1986. [Google Scholar] [CrossRef]
- Giurdanella, G.; Longo, A.; Distefano, A.; Olivieri, M.; Cristaldi, M.; Cosentino, A.; Agafonova, A.; Caporarello, N.; Lupo, G.; Anfuso, C.D. The Anti-Inflammatory Effect of the Β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells 2021, 11, 51. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Longo, A.; Distefano, A.; Amorini, A.M.; Salmeri, M.; Zanghì, G.; Giallongo, C.; Giurdanella, G.; Lupo, G. Uveal Melanoma Cells Elicit Retinal Pericyte Phenotypical and Biochemical Changes in an in Vitro Model of Coculture. Int. J. Mol. Sci. 2020, 21, 5557. [Google Scholar] [CrossRef]
- Mannino, G.; Longo, A.; Gennuso, F.; Anfuso, C.D.; Lupo, G.; Giurdanella, G.; Giuffrida, R.; Furno, D.L. Effects of High Glucose Concentration on Pericyte-Like Differentiated Human Adipose-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2021, 22, 4604. [Google Scholar] [CrossRef]
- Lupo, G.; Motta, C.; Salmeri, M.; Spina-Purrello, V.; Alberghina, M.; Anfuso, C.D. An in Vitro Retinoblastoma Human Triple Culture Model of Angiogenesis: A Modulatory Effect of TGF-β. Cancer Lett. 2014, 354, 181–188. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | PGE2 Release (pg/mL) ± SD | VEGF Release (pg/mL) ± SD |
|---|---|---|
| None | 78 ± 8.2 | 41.6 ± 3.8 |
| Dexamethasone 10 nM | 40.6 ± 5.3 * | 32.3 ± 3.1 * |
| Cisplatin 30 μM | 232.4 ± 21.6 * | 131.6 ± 11.4 * |
| Cisplatin 30 μM + Dexamethasone 10 nM | 108.5 9.1 § | 78.1 ±6.9 § |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anfuso, C.D.; Cosentino, A.; Agafonova, A.; Zappalà, A.; Giurdanella, G.; Trovato Salinaro, A.; Calabrese, V.; Lupo, G. Pericytes of Stria Vascularis Are Targets of Cisplatin-Induced Ototoxicity: New Insights into the Molecular Mechanisms Involved in Blood-Labyrinth Barrier Breakdown. Int. J. Mol. Sci. 2022, 23, 15790. https://doi.org/10.3390/ijms232415790
Anfuso CD, Cosentino A, Agafonova A, Zappalà A, Giurdanella G, Trovato Salinaro A, Calabrese V, Lupo G. Pericytes of Stria Vascularis Are Targets of Cisplatin-Induced Ototoxicity: New Insights into the Molecular Mechanisms Involved in Blood-Labyrinth Barrier Breakdown. International Journal of Molecular Sciences. 2022; 23(24):15790. https://doi.org/10.3390/ijms232415790
Chicago/Turabian StyleAnfuso, Carmelina Daniela, Alessia Cosentino, Aleksandra Agafonova, Agata Zappalà, Giovanni Giurdanella, Angela Trovato Salinaro, Vittorio Calabrese, and Gabriella Lupo. 2022. "Pericytes of Stria Vascularis Are Targets of Cisplatin-Induced Ototoxicity: New Insights into the Molecular Mechanisms Involved in Blood-Labyrinth Barrier Breakdown" International Journal of Molecular Sciences 23, no. 24: 15790. https://doi.org/10.3390/ijms232415790
APA StyleAnfuso, C. D., Cosentino, A., Agafonova, A., Zappalà, A., Giurdanella, G., Trovato Salinaro, A., Calabrese, V., & Lupo, G. (2022). Pericytes of Stria Vascularis Are Targets of Cisplatin-Induced Ototoxicity: New Insights into the Molecular Mechanisms Involved in Blood-Labyrinth Barrier Breakdown. International Journal of Molecular Sciences, 23(24), 15790. https://doi.org/10.3390/ijms232415790