
Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention

 , and
, and
Abstract
1. Introduction
2. Proteins Responsible for Neurodegenerative Diseases, Their Association with Cell Organelles and Involvement in Cellular Functions
2.1. Amyloid-β
2.2. Tau-Protein
2.3. α-Synuclein
2.4. Prions
2.5. TAR DNA-Binding Protein 43
2.6. Mutant Proteins in Polyglutamine Diseases
3. Huntingtin–Protein Interactions as the Basis of Intracellular Pathologies and Potential Target for Therapeutic Intervention in Huntington Disease
4. The Spectrum of Possible Cellular Pathologies in Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
| Disease | Involved Proteins | Cellular and Intracellular Disorders |
|---|---|---|
| Alzheimer’s disease (AD) | APP and Aβ Tau | Impaired of protein folding and degradation [138] |
| Lysosomal insufficiency (reviewed in [31]) | ||
| Mitochondrial dysfunction [29,30] | ||
| Violation of signaling pathways | ||
| Changes in protein expression [139,140] | ||
| Synapses damage [26,27] | ||
| Altered microtubule stabilization [141] | ||
| Parkinson’s disease (PD) | α-Synuclein Tau Parkin | Impaired of protein folding and degradation [142] |
| Mitochondrial dysfunction (reviewed in [143]) | ||
| Endoplasmic reticulum stress [144] | ||
| Cellular transport disorders (reviewed in [145]) | ||
| Amyotrophic lateral sclerosis | TDP-43 SOD1 FUS | Mitochondrial dysfunction [146,147] |
| Violation of axonal transport [148] | ||
| Changes in RNA metabolism [149] | ||
| Impaired protein degradation and autophagy [150] | ||
| Frontal temporal dementia | Tau TDP-43 FUS | Changes in gene expression [151] |
| Endosomal trafficking dysregulation [152] | ||
| Altered microtubule stabilization [153] | ||
| Impaired protein degradation and autophagy (reviewed in [154]) | ||
| Prion diseases | PrPC and PrPSc | Mitochondrial dysfunction [155,156] |
| Endoplasmic reticulum stress [156] | ||
| Impaired protein degradation [157] | ||
| Synapses damage [158] | ||
| Huntington’s disease (HD) | HTT | Violation of protein folding and degradation [159,160] |
| mRNA toxicity [161] | ||
| Accumulation of toxic polyglutamine fragments [160,162,163] | ||
| Cellular transport disorders (organelles, vesicles, neurotransmitters) [115,116,164] | ||
| Violation of autophagy [165,166,167] | ||
| Violation of signaling pathways [168,169] | ||
| Mitochondrial dysfunction [170,171] | ||
| Mitotic disorders [172] | ||
| Transcription disorders [163] | ||
| Violation of the primary cilia formation [130] |
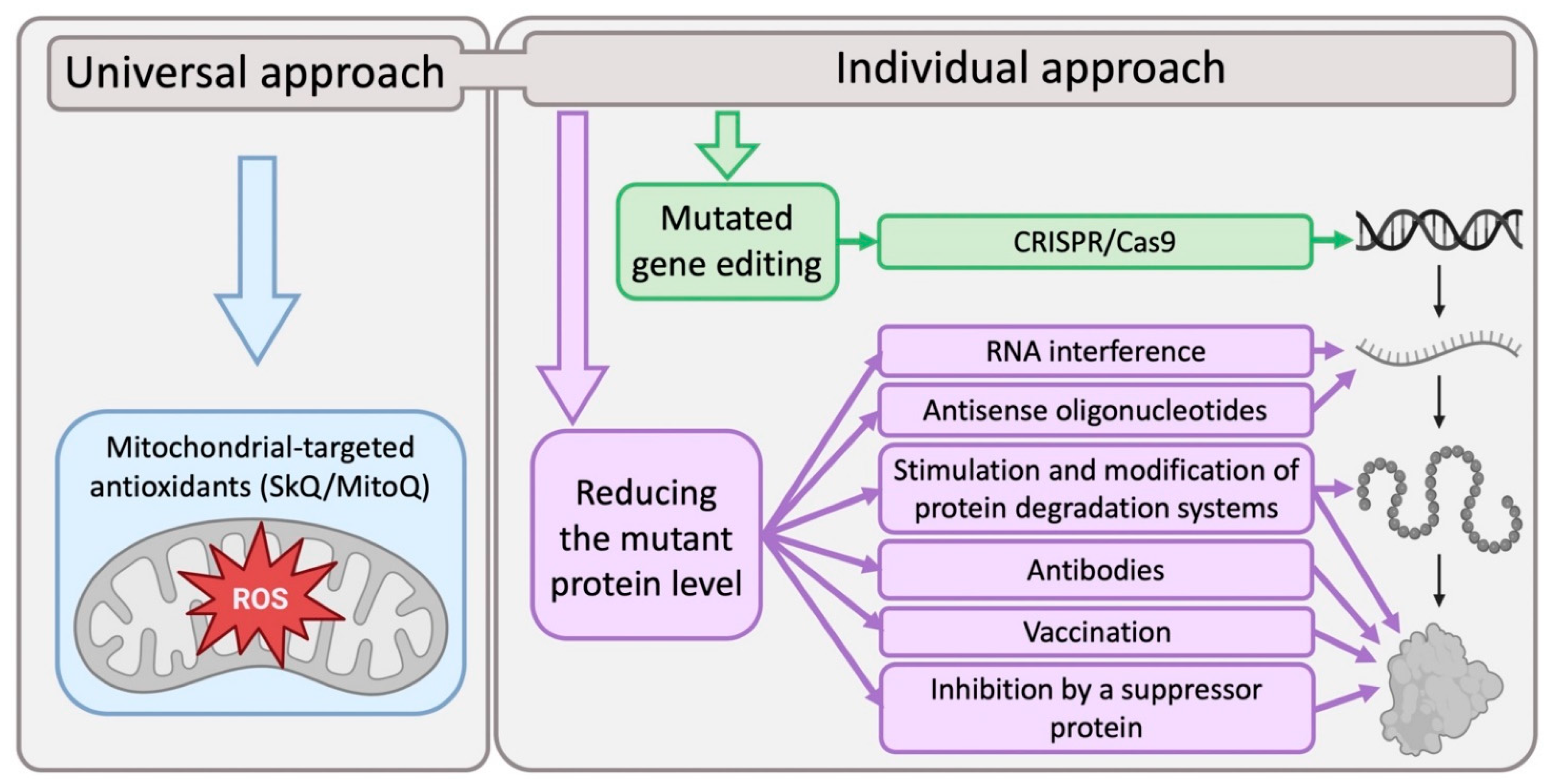
5. Modern Approaches to the Development of Methods for Neurodegenerative Diseases Treatment
6. Biomarkers for Early Detection of Neurodegenerative Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackenzie, I.R.A.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature for Neuropathologic Subtypes of Frontotemporal Lobar Degeneration: Consensus Recommendations. Acta Neuropathol. 2009, 117, 15–18. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and Nosology for Neuropathologic Subtypes of Frontotemporal Lobar Degeneration: An Update. Acta Neuropathol. 2010, 119, 1. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Invited Review: Neuropathology of Tauopathies: Principles and Practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Concepts and Classification of Neurodegenerative Diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 301–307. [Google Scholar]
- MacMillan, J.C.; Snell, R.G.; Tyler, A.; Houlihan, G.D.; Fenton, I.; Cheadle, J.P.; Lazarou, L.P.; Shaw, J.D.; Harper, P.S. Molecular Analysis and Clinical Correlations of the Huntington’s Disease Mutation. Lancet 1993, 342, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.J.; Kaleem, M.; Marlowe, L.; Pittman, A.M.; Lees, A.J.; Fung, H.C.; Duckworth, J.; Leung, D.; Gibson, A.; Morris, C.M.; et al. The H1c Haplotype at the MAPT Locus Is Associated with Alzheimer’s Disease. Hum. Mol. Genet. 2005, 14, 2399–2404. [Google Scholar] [CrossRef]
- Mattila, P.M.; Röyttä, M.; Torikka, H.; Dickson, D.W.; Rinne, J.O. Cortical Lewy Bodies and Alzheimer-Type Changes in Patients with Parkinson’s Disease. Acta Neuropathol. 1998, 95, 576–582. [Google Scholar] [CrossRef]
- Foster, E.R.; Campbell, M.C.; Burack, M.A.; Hartlein, J.; Flores, H.P.; Cairns, N.J.; Hershey, T.; Perlmutter, J.S. Amyloid Imaging of Lewy Body-Associated Disorders. Mov. Disord. 2010, 25, 2516–2523. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-Adenosylmethionine/Homocysteine Cycle Alterations Modify DNA Methylation Status with Consequent Deregulation of PS1 and BACE and Beta-Amyloid Production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Readhead, B.; Chen, K.; Su, Y.; Reiman, E.M.; Dudley, J.T. Longitudinal Data in Peripheral Blood Confirm That PM20D1 Is a Quantitative Trait Locus (QTL) for Alzheimer’s Disease and Implicate Its Dynamic Role in Disease Progression. Clin. Epigenetics 2020, 12, 189. [Google Scholar] [CrossRef]
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13, 639428. [Google Scholar] [CrossRef]
- Piller, C. Blots on a Field? Science 2022, 377, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Langui, D.; Ipsen, S.; Robakis, N.; Ulrich, J. Deposition of?/A4 Protein along Neuronal Plasma Membranes in Diffuse Senile Plaques. Acta Neuropathol. 1991, 83, 21–29. [Google Scholar] [CrossRef]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-Peptide Is Produced by Cultured Cells during Normal Metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schiossmacher, M.; Whaley, J.; Swindlehurst, C.; et al. Isolation and Quantification of Soluble Alzheimer’s β-Peptide from Biological Fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef]
- Busciglio, J.; Gabuzda, D.H.; Matsudaira, P.; Yankner, B.A. Generation of Beta-Amyloid in the Secretory Pathway in Neuronal and Nonneuronal Cells. Proc. Natl. Acad. Sci. USA 1993, 90, 2092–2096. [Google Scholar] [CrossRef]
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J.; et al. Secretion of β-Amyloid Precursor Protein Cleaved at the Amino Terminus of the β-Amyloid Peptide. Nature 1993, 361, 260–263. [Google Scholar] [CrossRef]
- Muller, U.C.; Zheng, H. Physiological Functions of APP Family Proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef]
- Baumkötter, F.; Wagner, K.; Eggert, S.; Wild, K.; Kins, S. Structural Aspects and Physiological Consequences of APP/APLP Trans-Dimerization. Exp. Brain Res. 2012, 217, 389–395. [Google Scholar] [CrossRef]
- Ho, A.; Südhof, T.C. Binding of F-Spondin to Amyloid-β Precursor Protein: A Candidate Amyloid-β Precursor Protein Ligand That Modulates Amyloid-β Precursor Protein Cleavage. Proc. Natl. Acad. Sci. USA 2004, 101, 2548–2553. [Google Scholar] [CrossRef]
- Park, J.H.; Gimbel, D.A.; GrandPre, T.; Lee, J.-K.; Kim, J.-E.; Li, W.; Lee, D.H.S.; Strittmatter, S.M. Alzheimer Precursor Protein Interaction with the Nogo-66 Receptor Reduces Amyloid-β Plaque Deposition. J. Neurosci. 2006, 26, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Cell Surface Amyloid β-Protein Precursor Colocalizes with Β1 Integrins at Substrate Contact Sites in Neural Cells. J. Neurosci. 1997, 17, 1004–1010. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Chen, A.C.; Chang, R.; Marquez, C.; Selkoe, D.J. Secreted APP Regulates the Function of Full-Length APP in Neurite Outgrowth through Interaction with Integrin Beta1. Neural Dev. 2008, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid-β Protein Forms Ion Channels: Implications for Alzheimer’s Disease Pathophysiology. FASEB J. 2001, 15, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-Amyloid Accumulation in APP Mutant Neurons Reduces PSD-95 and GluR1 in Synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid Inhibits Integrated Mitochondrial Respiration and Key Enzyme Activities. J. Neurochem. 2001, 80, 91–100. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria Are a Direct Site of Aβ Accumulation in Alzheimer’s Disease Neurons: Implications for Free Radical Generation and Oxidative Damage in Disease Progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M. Lysosomal System Pathways: Genes to Neurodegeneration in Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Haughey, N.J.; Nath, A.; Chan, S.L.; Borchard, A.C.; Rao, M.S.; Mattson, M.P. Disruption of Neurogenesis by Amyloid β-Peptide, and Perturbed Neural Progenitor Cell Homeostasis, in Models of Alzheimer’s Disease. J. Neurochem. 2002, 83, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yamamori, H.; Tatebayashi, Y.; Shafit-Zagardo, B.; Tanimukai, H.; Chen, S.; Iqbal, K.; Grundke-Iqbal, I. Failure of Neuronal Maturation in Alzheimer Disease Dentate Gyrus. J. Neuropathol. Exp. Neurol. 2008, 67, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of Tau, a Microtubule-Associated Protein That Induces Assembly of Microtubules from Purified Tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation Determines Two Distinct Species of Tau in the Central Nervous System. Cell Motil. Cytoskelet. 1987, 8, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of Tau Protein with the Dynactin Complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [PubMed]
- Selden, S.C.; Pollard, T.D. Phosphorylation of Microtubule-Associated Proteins Regulates Their Interaction with Actin Filaments. J. Biol. Chem. 1983, 258, 7064–7071. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, P.S.; Purich, D.L. Microtubule-Associated Protein Interactions with Actin Filaments: Evidence for Differential Behavior of Neuronal MAP-2 and Tau in the Presence of Phosphatidylinositol. Biochem. Biophys. Res. Commun. 1993, 190, 710–715. [Google Scholar] [CrossRef]
- Elie, A.; Prezel, E.; Guérin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau Co-Organizes Dynamic Microtubule and Actin Networks. Sci. Rep. 2015, 5, 9964. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of Tau with the Neural Plasma Membrane Mediated by Tau’s Amino-Terminal Projection Domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Arrasate, M.; Pérez, M.; Avila, J. Tau Dephosphorylation at Tau-1 Site Correlates with Its Association to Cell Membrane. Neurochem. Res. 2000, 25, 43–50. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation Regulates Tau Interactions with Src Homology 3 Domains of Phosphatidylinositol 3-Kinase, Phospholipase Cγ1, Grb2, and Src Family Kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Weissmann, C.; Golovyashkina, N.; Sebö-Lemke, Z.; Drewes, G.; Gerke, V.; Heinisch, J.J.; Brandt, R. The Frontotemporal Dementia Mutation R406W Blocks Tau’s Interaction with the Membrane in an Annexin A2–Dependent Manner. J. Cell Biol. 2011, 192, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau Interacts with Src-Family Non-Receptor Tyrosine Kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [CrossRef]
- Usardi, A.; Pooler, A.M.; Seereeram, A.; Reynolds, C.H.; Derkinderen, P.; Anderton, B.; Hanger, D.P.; Noble, W.; Williamson, R. Tyrosine Phosphorylation of Tau Regulates Its Interactions with Fyn SH2 Domains, but Not SH3 Domains, Altering the Cellular Localization of Tau. FEBS J. 2011, 278, 2927–2937. [Google Scholar] [CrossRef]
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear Magnetic Resonance Spectroscopy Characterization of Interaction of Tau with DNA and Its Regulation by Phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Wang, D.L.; Zhao, J.; Qu, M.H.; Zhou, X.H.; He, H.J.; He, R.Q. The Proline-Rich Domain and the Microtubule Binding Domain of Protein Tau Acting as RNA Binding Domains. Protein Pept. Lett. 2006, 13, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.-Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Conway, K.A.; Rochet, J.-C.; Bieganski, R.M.; Lansbury, P.T. Kinetic Stabilization of the α-Synuclein Protofibril by a Dopamine-α-Synuclein Adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.; Scheller, R. Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Nakai, M.; Fujita, M.; Waragai, M.; Sugama, S.; Wei, J.; Akatsu, H.; Ohtaka-Maruyama, C.; Okado, H.; Hashimoto, M. Expression of α-Synuclein, a Presynaptic Protein Implicated in Parkinson’s Disease, in Erythropoietic Lineage. Biochem. Biophys. Res. Commun. 2007, 358, 104–110. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. A-Synuclein Implicated in Parkinson’s Disease Is Present in Extracellular Biological Fluids, Including Human Plasma. FASEB J. 2003, 17, 1–16. [Google Scholar] [CrossRef]
- Lee, H.-J. Intravesicular Localization and Exocytosis of -Synuclein and Its Aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular Cloning of CDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Soper, J.H.; Kehm, V.; Burd, C.G.; Bankaitis, V.A.; Lee, V.M.-Y. Aggregation of α-Synuclein in S. Cerevisiae Is Associated with Defects in Endosomal Trafficking and Phospholipid Biosynthesis. J. Mol. Neurosci. 2011, 43, 391–405. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. Aβγ-Synuclein Triple Knockout Mice Reveal Age-Dependent Neuronal Dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins Regulate the Kinetics of Synaptic Vesicle Endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Role of α-Synuclein in Synaptic Glutamate Release. Neurobiol. Dis. 2007, 28, 83–89. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.H.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A Cellular Gene Encodes Scrapie PrP 27-30 Protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Basler, K.; Oesch, B.; Scott, M.; Westaway, D.; Wälchli, M.; Groth, D.F.; McKinley, M.P.; Prusiner, S.B.; Weissmann, C. Scrapie and Cellular PrP Isoforms Are Encoded by the Same Chromosomal Gene. Cell 1986, 46, 417–428. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Safar, J.; Roller, P.P.; Gajdusek, D.C.; Gibbs, C.J. Conformational Transitions, Dissociation, and Unfolding of Scrapie Amyloid (Prion) Protein. J. Biol. Chem. 1993, 268, 20276–20284. [Google Scholar] [CrossRef]
- Merz, P.A.; Somerville, R.A.; Wisniewski, H.M.; Iqbal, K. Abnormal Fibrils from Scrapie-Infected Brain. Acta Neuropathol. 1981, 54, 63–74. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; von Schroetter, C.; López García, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR Solution Structure of the Human Prion Protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef]
- Hay, B.; Prusiner, S.B.; Lingappa, V.R. Evidence for a Secretory Form of the Cellular Prion Protein. Biochemistry 1987, 26, 8110–8115. [Google Scholar] [CrossRef]
- Robertson, C.; Booth, S.A.; Beniac, D.R.; Coulthart, M.B.; Booth, T.F.; McNicol, A. Cellular Prion Protein Is Released on Exosomes from Activated Platelets. Blood 2006, 107, 3907–3911. [Google Scholar] [CrossRef]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal Cellular Prion Protein Drives Fibrillization of Amyloid Beta and Counteracts Amyloid Beta-Mediated Neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef]
- McLennan, N.F.; Rennison, K.A.; Bell, J.E.; Ironside, J.W. In Situ Hybridization Analysis of PrP MRNA in Human CNS Tissues. Neuropathol. Appl. Neurobiol. 2001, 27, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, P.; Bouzamondo-Bernstein, E.; Heinrich, C.; Prusiner, S.B.; DeArmond, S.J. Developmental Expression of PrP in the Post-Implantation Embryo. Brain Res. 2007, 1139, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Huckle, W.R.; Eyestone, W.H. Expression and Knockdown of Cellular Prion Protein (PrPC) in Differentiating Mouse Embryonic Stem Cells. Differentiation 2011, 81, 68–77. [Google Scholar] [CrossRef]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.-P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal Development and Behaviour of Mice Lacking the Neuronal Cell-Surface PrP Protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Baskakov, I.V. The Cellular Form of the Prion Protein Is Involved in Controlling Cell Cycle Dynamics, Self-Renewal, and the Fate of Human Embryonic Stem Cell Differentiation. J. Neurochem. 2013, 124, 310–322. [Google Scholar] [CrossRef]
- Prodromidou, K.; Papastefanaki, F.; Sklaviadis, T.; Matsas, R. Functional Cross-Talk Between the Cellular Prion Protein and the Neural Cell Adhesion Molecule Is Critical for Neuronal Differentiation of Neural Stem/Precursor Cells. Stem Cells 2014, 32, 1674–1687. [Google Scholar] [CrossRef]
- Halliez, S.; Martin-Lannerée, S.; Passet, B.; Hernandez-Rapp, J.; Castille, J.; Urien, C.; Chat, S.; Laude, H.; Vilotte, J.-L.; Mouillet-Richard, S.; et al. Prion Protein Localizes at the Ciliary Base during Neural and Cardiovascular Development and Its Depletion Affects α-Tubulin Post-Translational Modifications. Sci. Rep. 2015, 5, 17146. [Google Scholar] [CrossRef] [PubMed]
- Adle-Biassette, H.; Verney, C.; Peoc’h, K.; Dauge, M.-C.; Razavi, F.; Choudat, L.; Gressens, P.; Budka, H.; Henin, D. Immunohistochemical Expression of Prion Protein (PrPC) in the Human Forebrain During Development. J. Neuropathol. Exp. Neurol. 2006, 65, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.R.S.; Arantes, C.P.; Muras, A.G.; Nomizo, R.; Brentani, R.R.; Martins, V.R. Cellular Prion Protein Expression in Astrocytes Modulates Neuronal Survival and Differentiation. J. Neurochem. 2007, 103, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Skedsmo, F.S.; Malachin, G.; Våge, D.I.; Hammervold, M.M.; Salvesen, Ø.; Ersdal, C.; Ranheim, B.; Stafsnes, M.H.; Bartosova, Z.; Bruheim, P.; et al. Demyelinating Polyneuropathy in Goats Lacking Prion Protein. FASEB J. 2020, 34, 2359–2375. [Google Scholar] [CrossRef]
- Cashman, N.R.; Loertscher, R.; Nalbantoglu, J.; Shaw, I.; Kascsak, R.J.; Bolton, D.C.; Bendheim, P.E. Cellular Isoform of the Scrapie Agent Protein Participates in Lymphocyte Activation. Cell 1990, 61, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Burthem, J.; Urban, B.; Pain, A.; Roberts, D.J. The Normal Cellular Prion Protein Is Strongly Expressed by Myeloid Dendritic Cells. Blood 2001, 98, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular Prion Protein Controls Stem Cell-like Properties of Human Glioblastoma Tumor-Initiating Cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, C.W.; Han, Y.-S.; Kim, S.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.-J.; Lee, S.H. Melatonin and 5-Fluorouracil Co-Suppress Colon Cancer Stem Cells by Regulating Cellular Prion Protein-Oct4 Axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lin, C.-H.; Shih, N.-C.; Liu, H.-L.; Wang, W.-C.; Lin, K.-Y.; Liu, Z.-Y.; Tseng, Y.-J.; Chang, H.-K.; Lin, Y.-C.; et al. Cellular Prion Protein Transcriptionally Regulated by NFIL3 Enhances Lung Cancer Cell Lamellipodium Formation and Migration through JNK Signaling. Oncogene 2020, 39, 385–398. [Google Scholar] [CrossRef]
- Williams, W.M.; Stadtman, E.R.; Moskovitz, J. Ageing and Exposure to Oxidative Stress in Vivo Differentially Affect Cellular Levels of PrP c in Mouse Cerebral Microvessels and Brain Parenchyma. Neuropathol. Appl. Neurobiol. 2004, 30, 161–168. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Legname, G.; Benetti, F. In Absence of the Cellular Prion Protein, Alterations in Copper Metabolism and Copper-Dependent Oxidase Activity Affect Iron Distribution. Front. Neurosci. 2016, 10, 437. [Google Scholar] [CrossRef]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The Cellular Prion Protein Traps Alzheimer’s Aβ in an Oligomeric Form and Disassembles Amyloid Fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef]
- Ulbrich, S.; Janning, P.; Seidel, R.; Matschke, J.; Gonsberg, A.; Jung, S.; Glatzel, M.; Engelhard, M.; Winklhofer, K.F.; Tatzelt, J. Alterations in the Brain Interactome of the Intrinsically Disordered N-Terminal Domain of the Cellular Prion Protein (PrPC) in Alzheimer’s Disease. PLoS ONE 2018, 13, e0197659. [Google Scholar] [CrossRef]
- Buratti, E. Multiple Roles of TDP-43 in Gene Expression, Splicing Regulation, and Human Disease. Front. Biosci. 2008, 13, 867. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 Regulates Retinoblastoma Protein Phosphorylation through the Repression of Cyclin-Dependent Kinase 6 Expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Amador-Ortiz, C.; Lin, W.-L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 Immunoreactivity in Hippocampal Sclerosis and Alzheimer’s Disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B.; et al. Co-Morbidity of TDP-43 Proteinopathy in Lewy Body Related Diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Akiyama, H.; Nonaka, T.; Mori, H.; Hashimoto, T.; Yamazaki, M.; Oyanagi, K. TDP-43 Is Deposited in the Guam Parkinsonism-Dementia Complex Brains. Brain 2007, 130, 1386–1394. [Google Scholar] [CrossRef]
- Dammer, E.B.; Fallini, C.; Gozal, Y.M.; Duong, D.M.; Rossoll, W.; Xu, P.; Lah, J.J.; Levey, A.I.; Peng, J.; Bassell, G.J.; et al. Coaggregation of RNA-Binding Proteins in a Model of TDP-43 Proteinopathy with Selective RGG Motif Methylation and a Role for RRM1 Ubiquitination. PLoS ONE 2012, 7, e38658. [Google Scholar] [CrossRef]
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; et al. The RNA-Binding Motif 45 (RBM45) Protein Accumulates in Inclusion Bodies in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Lobar Degeneration with TDP-43 Inclusions (FTLD-TDP) Patients. Acta Neuropathol. 2012, 124, 717–732. [Google Scholar] [CrossRef]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 Gene Reveals a Highly Unstable CAG Repeat Expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG Expansions in a Novel Gene for Machado-Joseph Disease at Chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T.; et al. Unstable Expansion of CAG Repeat in Hereditary Dentatorubral–Pallidoluysian Atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K. SCA17, a Novel Autosomal Dominant Cerebellar Ataxia Caused by an Expanded Polyglutamine in TATA-Binding Protein. Hum. Mol. Genet. 2001, 10, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Chung, M.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.W.; Zoghbi, H.Y. Expansion of an Unstable Trinucleotide CAG Repeat in Spinocerebellar Ataxia Type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.-M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.-N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate Expansion of a Normally Biallelic Trinucleotide Repeat in Spinocerebellar Ataxia Type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal Dominant Cerebellar Ataxia (SCA6) Associated with Small Polyglutamine Expansions in the A1A-Voltage-Dependent Calcium Channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen Receptor Gene Mutations in X-Linked Spinal and Bulbar Muscular Atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Dewan, R.; Chia, R.; Ding, J.; Hickman, R.A.; Stein, T.D.; Abramzon, Y.; Ahmed, S.; Sabir, M.S.; Portley, M.K.; Tucci, A.; et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 2021, 109, 448–460.e4. [Google Scholar] [CrossRef]
- Manini, A.; Gagliardi, D.; Meneri, M.; Antognozzi, S.; del Bo, R.; Scaglione, C.; Comi, G.P.; Corti, S.; Ronchi, D. Analysis of HTT CAG Repeat Expansion in Italian Patients with Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2022, 9, 1820–1825. [Google Scholar] [CrossRef]
- Coudert, L.; Nonaka, T.; Bernard, E.; Hasegawa, M.; Schaeffer, L.; Leblanc, P. Phosphorylated and Aggregated TDP-43 with Seeding Properties Are Induced upon Mutant Huntingtin (mHTT) Polyglutamine Expression in Human Cellular Models. Cell. Mol. Life Sci. 2019, 76, 2615–2632. [Google Scholar] [CrossRef]
- Hickman, R.A.; Dewan, R.; Cortes, E.; Traynor, B.J.; Marder, K.; Vonsattel, J.-P. Amyotrophic Lateral Sclerosis Is Over-Represented in Two Huntington’s Disease Brain Bank Cohorts: Further Evidence to Support Genetic Pleiotropy of Pathogenic HTT Gene Expansion. Acta Neuropathol. 2022, 143, 105–108. [Google Scholar] [CrossRef]
- Sousa, C.M.; Humbert, S. Huntingtin: Here, There, Everywhere! J. Huntingt. Dis. 2013, 2, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A Polymorphic DNA Marker Genetically Linked to Huntington’s Disease. Nature 1983, 307, 234–238. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s Disease Genetics. NeuroRX 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; de Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Zajac, A.L.; Tokito, M.; Holzbaur, E.L.F. Huntingtin Coordinates the Dynein-Mediated Dynamic Positioning of Endosomes and Lysosomes. Mol. Biol. Cell 2010, 22, 478–492. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARs to Synapses Is Mediated by HAP1-KIF5 and Disrupted by Mutant Huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin Alters Retrograde Transport of TrkB Receptors in Striatal Dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef]
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.; Holzbaur, E.L.F.; Ross, C.A. Huntingtin-Associated Protein 1 (HAP1) Interacts with the P150Glued Bubunit of Dynactin. Hum. Mol. 1997, 6, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin Facilitates Dynein/Dynactin-Mediated Vesicle Transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007, 3, 689–708. [Google Scholar] [CrossRef]
- El-Daher, M.-T.; Hangen, E.; Bruyere, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin Proteolysis Releases Non-PolyQ Fragments That Cause Toxicity through Dynamin 1 Dysregulation. EMBO J. 2015, 34, 2255–2271. [Google Scholar] [CrossRef] [PubMed]
- Taran, A.S.; Shuvalova, L.D.; Lagarkova, M.A.; Alieva, I.B. Huntington’s Disease-An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells 2020, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Taran, A.; Belikova, L.; Lavrushkina, S.; Bogomazova, A.; Lagarkova, M.; Alieva, I. Microtubule Plus-End Dynamics Visualization in Huntington’s Disease Model Based on Human Primary Skin Fibroblasts. J. Vis. Exp. 2022, 2022, e62963. [Google Scholar] [CrossRef]
- Steffan, J.S. Does Huntingtin Play a Role in Selective Macroautophagy? Cell Cycle 2010, 9, 3401–3413. [Google Scholar] [CrossRef]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington Disease and Huntingtin in Autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Gusella, J.F. The Predominantly HEAT-like Motif Structure of Huntingtin and Its Association and Coincident Nuclear Entry with Dorsal, an NF- ΚB/Rel/Dorsal Family Transcription Factor. BMC Neurosci. 2002, 3, 15. [Google Scholar] [CrossRef]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.-H.J. Huntingtin Modulates Transcription, Occupies Gene Promoters In Vivo, and Binds Directly to DNA in a Polyglutamine-Dependent Manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.-L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin Is Required for Mitotic Spindle Orientation and Mammalian Neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef]
- Keryer, G.; Dragatsis, I.; Saudou, F.; Keryer, G.; Pineda, J.R.; Liot, G.; Kim, J.; Dietrich, P.; Ferrante, R.J.; Dragatsis, I.; et al. Ciliogenesis Is Regulated by a Huntingtin-HAP1-PCM1 Pathway and Is Altered in Huntington Disease. J. Clin. Investig. 2011, 121, 4372–4382. [Google Scholar] [CrossRef]
- Haremaki, T.; Deglincerti, A.; Brivanlou, A.H. Huntingtin Is Required for Ciliogenesis and Neurogenesis during Early Xenopus Development. Dev. Biol. 2015, 408, 305–315. [Google Scholar] [CrossRef]
- Karam, A.; Tebbe, L.; Weber, C.; Messaddeq, N.; Morlé, L.; Kessler, P.; Wolfrum, U.; Trottier, Y. A Novel Function of Huntingtin in the Cilium and Retinal Ciliopathy in Huntington’s Disease Mice. Neurobiol. Dis. 2015, 80, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Culver, B.P.; Savas, J.N.; Park, S.K.; Choi, J.H.; Zheng, S.; Zeitlin, S.O.; Yates, J.R.; Tanese, N. Proteomic Analysis of Wild-Type and Mutant Huntingtin-Associated Proteins in Mouse Brains Identifies Unique Interactions and Involvement in Protein Synthesis. J. Biol. Chem. 2012, 287, 21599–21614. [Google Scholar] [CrossRef]
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A Large Scale Huntingtin Protein Interaction Network Implicates RHO GTPase Signaling Pathways in Huntington Disease. J. Biol. Chem. 2014, 289, 6709–6726. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H.; Vonsattel, J.P.; Paskevich, P.A.; Kiely, D.K.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P.; Bird, E.D. Decreased Neuronal and Increased Oligodendroglial Densities in Huntington’s Disease Caudate Nucleus. J. Neuropathol. Exp. Neurol. 1991, 50, 729–742. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The Role of Glial Reaction and Inflammation in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2006, 991, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Powers, E.T.; Nieva, J.; Huff, M.E.; Dendle, M.A.; Bieschke, J.; Glabe, C.G.; Eschenmoser, A.; Wentworth, P.; Lerner, R.A.; et al. Metabolite-Initiated Protein Misfolding May Trigger Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 4752–4757. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Matarin, M.; Salih, D.A.; Yasvoina, M.; Cummings, D.M.; Guelfi, S.; Liu, W.; Nahaboo Solim, M.A.; Moens, T.G.; Paublete, R.M.; Ali, S.S.; et al. A Genome-Wide Gene-Expression Analysis and Database in Transgenic Mice during Development of Amyloid or Tau Pathology. Cell Rep. 2015, 10, 633–644. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal Function Is Impaired in Substantia Nigra in Parkinson’s Disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial Dysfunction in Neurodegenerative Diseases and Drug Targets via Apoptotic Signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J. Alpha-Synuclein Induces the Unfolded Protein Response in Parkinson’s Disease SNCA Triplication IPSC-Derived Neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in Trafficking Bridge Parkinson’s Disease Pathology and Genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An Adverse Property of a Familial ALS-Linked SOD1 Mutation Causes Motor Neuron Disease Characterized by Vacuolar Degeneration of Mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients With Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Impairment of Fast Axonal Transport in the Proximal Axons of Anterior Horn Neurons in Amyotrophic Lateral Sclerosis. Neurology 1996, 47, 535–540. [Google Scholar] [CrossRef]
- Lederer, C.W.; Torrisi, A.; Pantelidou, M.; Santama, N.; Cavallaro, S. Pathways and Genes Differentially Expressed in the Motor Cortex of Patients with Sporadic Amyotrophic Lateral Sclerosis. BMC Genom. 2007, 8, 26. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Chen-Plotkin, A.S.; Geser, F.; Plotkin, J.B.; Clark, C.M.; Kwong, L.K.; Yuan, W.; Grossman, M.; van Deerlin, V.M.; Trojanowski, J.Q.; Lee, V.M.-Y. Variations in the Progranulin Gene Affect Global Gene Expression in Frontotemporal Lobar Degeneration. Hum. Mol. Genet. 2008, 17, 1349–1362. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, Implicated in Amytrophic Lateral Sclerosis and Frontotemporal Dementia, Regulates Endosomal Trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Zheng-Fischhöfer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.-M.; Mandelkow, E. Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias. Biochemistry 2000, 39, 11714–11721. [Google Scholar] [CrossRef]
- Götzl, J.K.; Lang, C.M.; Haass, C.; Capell, A. Impaired Protein Degradation in FTLD and Related Disorders. Ageing Res. Rev. 2016, 32, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski-Treska, J.; Grant, N.J.; Ulrich, G.; Corrotte, M.; Bailly, Y.; Haeberle, A.-M.; Chasserot-Golaz, S.; Bader, M.-F. Fibrillar Prion Peptide (106-126) and Scrapie Prion Protein Hamper Phagocytosis in Microglia. Glia 2004, 46, 101–115. [Google Scholar] [CrossRef]
- Hetz, C. Caspase-12 and Endoplasmic Reticulum Stress Mediate Neurotoxicity of Pathological Prion Protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.B.; Menéndez-Benito, V.; Dantuma, N.P.; et al. Disease-Associated Prion Protein Oligomers Inhibit the 26S Proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Brown, D.; Jeffrey, M.; Fraser, J.R. Dendritic and Synaptic Alterations of Hippocampal Pyramidal Neurones in Scrapie-Infected Mice. Neuropathol. Appl. Neurobiol. 2000, 26, 143–149. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of Neuronal Intranuclear Inclusions Underlies the Neurological Dysfunction in Mice Transgenic for the HD Mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Bañez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzmán, M.; Estivill, X.; Martí, E. A Pathogenic Mechanism in Huntington’s Disease Involves Small CAG-Repeated RNAs with Neurotoxic Activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin Acts in the Nucleus to Induce Apoptosis but Death Does Not Correlate with the Formation of Intranuclear Inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Valor, L.M. Transcription, Epigenetics and Ameliorative Strategies in Huntington’s Disease: A Genome-Wide Perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Zala, D.; Hinckelmann, M.V.; Saudou, F. Huntingtin’s Function in Axonal Transport Is Conserved in Drosophila Melanogaster. PLoS ONE 2013, 8, e60162. [Google Scholar] [CrossRef]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castaño, J.G.; Aronin, N.; DiFiglia, M. Huntingtin Expression Stimulates Endosomal–Lysosomal Activity, Endosome Tubulation, and Autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of MTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo Recognition Failure Is Responsible for Inefficient Autophagy in Huntington’s Disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Rigamonti, D.; Sipione, S.; Goffredo, D.; Zuccato, C.; Fossale, E.; Cattaneo, E. Huntingtin’s Neuroprotective Activity Occurs via Inhibition of Procaspase-9 Processing. J. Biol. Chem. 2001, 276, 14545–14548. [Google Scholar] [CrossRef]
- Zhang, Y.; Leavitt, B.R.; van Raamsdonk, J.M.; Dragatsis, I.; Goldowitz, D.; MacDonald, M.E.; Hayden, M.R.; Friedlander, R.M. Huntingtin Inhibits Caspase-3 Activation. EMBO J. 2006, 25, 5896–5906. [Google Scholar] [CrossRef]
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on brain biopsies of patients with huntingtonʼs chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Macgarvey, U.; Baik, M.J.; Berger, S.C.; Muquit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative Damage and Metabolic Dysfunction in Huntington’s Disease: Selective Vulnerability of the Basal Ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Sathasivam, K. Centrosome Disorganization in Fibroblast Cultures Derived from R6/2 Huntington’s Disease (HD) Transgenic Mice and HD Patients. Hum. Mol. Genet. 2001, 10, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic Aβ Peptides Increase Oxidative Stress in Vivo through NMDA-Receptor and Nitric-Oxide-Synthase Mechanisms, and Inhibit Complex IV Activity and Induce a Mitochondrial Permeability Transition in Vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. β-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, D.; Smith, M.A.; Richey, P.L.; Vinters, H.V.; Perry, G. Filament Heterogeneity within the Dystrophic Neurites of Senile Plaques Suggests Blockage of Fast Axonal Transport in Alzheimer’s Disease. Acta Neuropathol. 1996, 91, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial Hyperpolarization in IPSC-Derived Neurons from Patients of FTDP-17 with 10+16 MAPT Mutation Leads to Oxidative Stress and Neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical Reporters for the Conformation of α-Synuclein Reveal a Specific Interaction with Mitochondria. J. Neurosci. 2008, 28, 12305. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-Synuclein Oligomers Promote Complex I-Dependent, Ca2+-Induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef][Green Version]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.-C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.-M.; Hirsch, E.; Hantraye, P.; et al. Involvement of Mitochondrial Complex II Defects in Neuronal Death Produced by N-Terminus Fragment of Mutated Huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium Homeostasis and Mitochondrial Dysfunction in Striatal Neurons of Huntington Disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Plowey, E.D.; Wang, Y.; Patel, V.; Jordan-Sciutto, K.L. Location, Location, Location: Altered Transcription Factor Trafficking in Neurodegeneration. J. Neuropathol. Exp. Neurol. 2007, 66, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Noble, W.; Tartaglia, G.G.; Buckley, N.J. Neurodegeneration as an RNA Disorder. Prog. Neurobiol. 2012, 99, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Krach, F.; Stemick, J.; Boerstler, T.; Weiss, A.; Lingos, I.; Reischl, S.; Meixner, H.; Ploetz, S.; Farrell, M.; Hehr, U.; et al. An Alternative Splicing Modulator Decreases Mutant HTT and Improves the Molecular Fingerprint in Huntington’s Disease Patient Neurons. Nat. Commun. 2022, 13, 6797. [Google Scholar] [CrossRef]
- Johnson, M.D.; Yu, L.-R.; Conrads, T.P.; Kinoshita, Y.; Uo, T.; McBee, J.K.; Veenstra, T.D.; Morrison, R.S. The Proteomics of Neurodegeneration. Am. J. Pharm. 2005, 5, 259–270. [Google Scholar] [CrossRef]
- Skariah, G.; Todd, P.K. Translational Control in Aging and Neurodegeneration. WIREs RNA 2021, 12, e1628. [Google Scholar] [CrossRef]
- Shinn, L.J.; Lagalwar, S. Treating Neurodegenerative Disease with Antioxidants: Efficacy of the Bioactive Phenol Resveratrol and Mitochondrial-Targeted MitoQ and SkQ. Antioxidants 2021, 10, 573. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.J.; Murphy, M.P.; Taylor, K.M. A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson’s Disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Song, G.; Yang, H.; Shen, N.; Pham, P.; Brown, B.; Lin, X.; Hong, Y.; Sinu, P.; Cai, J.; Li, X.; et al. An Immunomodulatory Therapeutic Vaccine Targeting Oligomeric Amyloid-β1. J. Alzheimer’s Dis. 2020, 77, 1639–1653. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s Disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.-M. Permanent Inactivation of Huntington’s Disease Mutation by Personalized Allele-Specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA Interference Improves Motor and Neuropathological Abnormalities in a Huntington’s Disease Mouse Model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic Silencing of Mutant Huntingtin with SiRNA Attenuates Striatal and Cortical Neuropathology and Behavioral Deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Gatsonis, C.; Apgar, C.; Chaudhary, K.; Gareen, I.; Hanna, L.; Hendrix, J.; Hillner, B.E.; Olson, C.; Lesman-Segev, O.H.; et al. Association of Amyloid Positron Emission Tomography With Subsequent Change in Clinical Management Among Medicare Beneficiaries With Mild Cognitive Impairment or Dementia. JAMA 2019, 321, 1286. [Google Scholar] [CrossRef]
- Palmqvist, S.; Mattsson, N.; Hansson, O. Cerebrospinal Fluid Analysis Detects Cerebral Amyloid-β Accumulation Earlier than Positron Emission Tomography. Brain 2016, 139, 1226–1236. [Google Scholar] [CrossRef]
- Mattsson, N.; Lönneborg, A.; Boccardi, M.; Blennow, K.; Hansson, O. Clinical Validity of Cerebrospinal Fluid Aβ42, Tau, and Phospho-Tau as Biomarkers for Alzheimer’s Disease in the Context of a Structured 5-Phase Development Framework. Neurobiol. Aging 2017, 52, 196–213. [Google Scholar] [CrossRef]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and Blood Biomarkers for Parkinson’s Disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orrú, C.D.; Tatsuoka, C.; et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2021, 78, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Ma, J.-Y.; Liu, X.-Y.; Zhang, S.; Yu, Z.; Fan, D.-S. Promising Application of a New Ulnar Nerve Compound Muscle Action Potential Measurement Montage in Amyotrophic Lateral Sclerosis: A Prospective Cross-Sectional Study. Neural Regen. Res. 2023, 18, 908. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Churkina, A.S.; Shakhov, A.S.; Kotlobay, A.A.; Alieva, I.B. Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 15533. https://doi.org/10.3390/ijms232415533
Churkina AS, Shakhov AS, Kotlobay AA, Alieva IB. Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention. International Journal of Molecular Sciences. 2022; 23(24):15533. https://doi.org/10.3390/ijms232415533
Chicago/Turabian StyleChurkina (Taran), Aleksandra S., Anton S. Shakhov, Anatoly A. Kotlobay, and Irina B. Alieva. 2022. "Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention" International Journal of Molecular Sciences 23, no. 24: 15533. https://doi.org/10.3390/ijms232415533
APA StyleChurkina, A. S., Shakhov, A. S., Kotlobay, A. A., & Alieva, I. B. (2022). Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention. International Journal of Molecular Sciences, 23(24), 15533. https://doi.org/10.3390/ijms232415533