
Alzheimer’s Disease: Treatment Strategies and Their Limitations

 ,
,  ,
,  , ,
, , 
 and
and 
Abstract
1. Introduction
2. Understanding Clinical Spectrum of AD
2.1. Description
2.1.1. Clinical
2.1.2. Pathophysiology
2.2. Diagnostic Criteria for AD
2.2.1. Criteria of the National Institute of Aging and the Alzheimer’s Association (NIA-AA)
2.2.2. Specific AD Biomarkers
2.3. The Different Stages of the Sporadic form of AD: The Alzheimer’s Spectrum
2.3.1. The Early Asymptomatic Stage: Preclinical Stage
2.3.2. The Early Symptomatic Stage: Amnesic Mild Cognitive Impairment
2.3.3. AD
2.4. Risk Factors of SAD
2.4.1. Non-Modifiable Risk Factors
Age
Genetic Risk Factor
Gender
2.4.2. Modifiable Risk Factors
Metabolic Disorders and Dyslipidemia
Other Risk Factors
3. Research Models Used for AD
3.1. 2D In Vitro Models of Alzheimer’s Disease
3.2. 3D Models of Alzheimer’s Disease
3.3. In Vivo Animal Models of AD
4. Current Treatments and Prevention Strategies for Alzheimer’s Disease
4.1. Drug Treatments
Description
4.2. Non-Pharmacological Therapies
4.3. Prevention Strategies for Alzheimer’s Disease
4.3.1. Dietary Intervention
Fatty Acids: Omega 3, 6
Docosahexaenoic Acid
5. BBB and Administration Strategies for Alzheimer’s Disease Treatment
5.1. BBB and Soft Nanoparticles
5.1.1. Liposomes
5.1.2. Exosomes
5.2. Oral Administration
5.3. Intravenous and Intracerebral Administration
5.4. Intranasal Administration
5.5. Novel Administration Strategies
5.5.1. Ultrasound and Electromagnetism
5.5.2. Transdermal Delivery Systems via Microneedles
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| Aβ | Beta-amyloid peptide |
| AβO | Aβ oligomers |
| AD | Alzheimer’s disease |
| ADAD | Autosomal-dominant Alzheimer’s disease |
| ALA | Alpha linolenic acid |
| aMCI | Amnesic mild cognitive impairment |
| APOE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| BBB | Blood–brain barrier |
| BPSD | Behavioral and psychological symptoms of dementia |
| CBS | Cognitive-behavioral syndrome |
| CSF | Cerebrospinal fluid |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| DHA | Docosahexaenoic acid |
| ECM | Extracellular matrix |
| EPA | Eicosapentaenoic acid |
| FA | Fatty acid |
| FDG-PET | Fluorodeoxyglucose positron emission tomography |
| GFP | Green Fluorescent Protein |
| HSV-1 | Herpes simplex virus type I |
| IN | Intranasal |
| iPSC | Pluripotent stem cell |
| LA | Linoleic acid |
| LC-PUFA | Long-chain polyunsaturated fatty acid |
| Lf | Lactoferrin |
| MCI | Mild cognitive impairment |
| MOR | Opioid receptor mu |
| MRI | Magnetic resonance imaging |
| n-3 | Omega-3 |
| n-3 PUFA | Omega-3 polyunsaturated fatty acid |
| n-6 | Omega 6 |
| ND | Neurodegenerative disease |
| NFT | Neurofibrillary tangles |
| NHP | Non-human primate |
| NIA-AA | National Institute of Aging and the Alzheimer’s Association |
| NL | Nanoliposome |
| NMDA | N-methyl-D-aspartate |
| NP | Nanoparticle |
| NPC | Neural progenitor cell |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PET | Positron emission tomography |
| PITRM1 | Pitrilysin metallopeptidase 1 |
| PL | Phospholipid |
| PLGA | Poly(lactic-co-glycolic acid) |
| PS | Phosphatidylserine |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| PUFA | Polyunsaturated fatty acid |
| ROS | Reactive oxygen species |
| RVG | Rabies virus glycoprotein |
| SAD | Sporadic Alzheimer Disease |
| SEM | Scanning electron microscopy |
| Tau | Tubule Associated Unit |
| Tf | Transferrin |
| TfR | Transferrin receptor |
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s Disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Prince, M.J. World Alzheimer Report 2015: The Global Impact of Dementia. Available online: https://www.alz.co.uk/research/world-report-2015 (accessed on 7 March 2019).
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tan, L.; Liu, J.; Yu, J.-T. The Essential Role of Soluble Aβ Oligomers in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 1905–1924. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-Protein Oligomers and Alzheimer’s Disease. Alzheimer’s Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef]
- Mucke, L. Neuroscience: Alzheimer’s Disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and Away from Alzheimer’s Disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Vickers, J.C.; Mitew, S.; Woodhouse, A.; Fernandez-Martos, C.M.; Kirkcaldie, M.T.; Canty, A.J.; McCormack, G.H.; King, A.E. Defining the Earliest Pathological Changes of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β Deposition, Neurodegeneration, and Cognitive Decline in Sporadic Alzheimer’s Disease: A Prospective Cohort Study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 978-0-89042-554-1. [Google Scholar]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New Approaches for the Treatment of Alzheimer’s Disease. Bioorganic Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef]
- Cummings, J.; Fox, N. Defining Disease Modifying Therapy for Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 109. [Google Scholar] [CrossRef]
- Andrieu, S.; Coley, N.; Lovestone, S.; Aisen, P.S.; Vellas, B. Prevention of Sporadic Alzheimer’s Disease: Lessons Learned from Clinical Trials and Future Directions. Lancet Neurol. 2015, 14, 926–944. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for Primary Prevention of Alzheimer’s Disease: An Analysis of Population-Based Data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia Prevention, Intervention, and Care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s Disease: Definition, Natural History, and Diagnostic Criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Spector, A.A. Essentiality of Fatty Acids. Lipids 1999, 34, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Essential Fatty Acids in Growth and Development. Prog. Lipid Res. 1991, 30, 39–103. [Google Scholar] [CrossRef]
- Kawakita, E.; Hashimoto, M.; Shido, O. Docosahexaenoic Acid Promotes Neurogenesis in Vitro and in Vivo. Neuroscience 2006, 139, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, L.A.; Farooqui, A.A. Docosahexaenoic Acid in the Diet: Its Importance in Maintenance and Restoration of Neural Membrane Function. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 361–372. [Google Scholar] [CrossRef]
- Lu, C.-T.; Zhao, Y.-Z.; Wong, H.L.; Cai, J.; Peng, L.; Tian, X.-Q. Current Approaches to Enhance CNS Delivery of Drugs across the Brain Barriers. Int. J. Nanomed. 2014, 9, 2241. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Preda, M.D.; Niculescu, A.-G.; Vladâcenco, O.; Radu, C.I.; Grumezescu, A.M.; Teleanu, D.M. Current Strategies to Enhance Delivery of Drugs across the Blood–Brain Barrier. Pharmaceutics 2022, 14, 987. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood-Brain Barrier to Treat Neurodegenerative Diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Poudel, P.; Park, S. Recent Advances in the Treatment of Alzheimer’s Disease Using Nanoparticle-Based Drug Delivery Systems. Pharmaceutics 2022, 14, 835. [Google Scholar] [CrossRef]
- Cano, A.; Turowski, P.; Ettcheto, M.; Duskey, J.T.; Tosi, G.; Sánchez-López, E.; García, M.L.; Camins, A.; Souto, E.B.; Ruiz, A.; et al. Nanomedicine-Based Technologies and Novel Biomarkers for the Diagnosis and Treatment of Alzheimer’s Disease: From Current to Future Challenges. J. Nanobiotechnol. 2021, 19, 122. [Google Scholar] [CrossRef]
- Agrawal, M.; Ajazuddin; Tripathi, D.K.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Mourtas, S.; Hammarlund-Udenaes, M.; Alexander, A. Recent Advancements in Liposomes Targeting Strategies to Cross Blood-Brain Barrier (BBB) for the Treatment of Alzheimer’s Disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Jayant, R.D.; Bhardwaj, V.; Nair, M. Personalized Nanomedicine for CNS Diseases. Drug Discov. Today 2018, 23, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.; Abramovitz, L.; Peer, D. Precision Nanomedicine in Neurodegenerative Diseases. ACS Nano 2014, 8, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.; Gamarra, L. Getting into the Brain: Liposome-Based Strategies for Effective Drug Delivery across the Blood-Brain Barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef]
- Ross, C.; Taylor, M.; Fullwood, N.; Allsop, D. Liposome Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2018, 13, 8507–8522. [Google Scholar] [CrossRef]
- Maherani, B.; Arab-Tehrany, E.; Mozafari, M.R.; Gaiani, C.; Linder, M. Liposomes: A Review of Manufacturing Techniques and Targeting Strategies. Available online: http://www.eurekaselect.com/73978/article (accessed on 7 March 2019).
- Khorasani, S.; Danaei, M.; Mozafari, M.R. Nanoliposome Technology for the Food and Nutraceutical Industries. Trends Food Sci. Technol. 2018, 79, 106–115. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent Advances with Liposomes as Pharmaceutical Carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Sharma, D.; Ali, A.A.E.; Trivedi, L.R. An Updated Review On:Liposomes as Drug Delivery System. PharmaTutor 2018, 6, 50. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Kales, H.C.; Gitlin, L.N.; Lyketsos, C.G. Assessment and Management of Behavioral and Psychological Symptoms of Dementia. BMJ 2015, 350, h369. [Google Scholar] [CrossRef]
- Lanctôt, K.L.; Amatniek, J.; Ancoli-Israel, S.; Arnold, S.E.; Ballard, C.; Cohen-Mansfield, J.; Ismail, Z.; Lyketsos, C.; Miller, D.S.; Musiek, E.; et al. Neuropsychiatric Signs and Symptoms of Alzheimer’s Disease: New Treatment Paradigms. Alzheimer’s Dement. 2017, 3, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Cerejeira, J.; Lagarto, L.; Mukaetova-Ladinska, E.B. Behavioral and Psychological Symptoms of Dementia. Front. Neur. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Finkel, S.I.; Costa e Silva, J.; Cohen, G.; Miller, S.; Sartorius, N. Behavioral and Psychological Signs and Symptoms of Dementia: A Consensus Statement on Current Knowledge and Implications for Research and Treatment. Int. Psychogeriatr. 1996, 8 (Suppl. 3), 497–500. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Allinquant, B.; Clamagirand, C.; Potier, M.-C. Role of Cholesterol Metabolism in the Pathogenesis of Alzheimer’s Disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β Oligomers in Alzheimer’s Disease Pathogenesis, Treatment, and Diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid Precursor Protein and Endosomal-lysosomal Dysfunction in Alzheimer’s Disease: Inseparable Partners in a Multifactorial Disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J.; Hone, E.; Foster, J.K.; Sünram-Lea, S.I.; Gnjec, A.; Fuller, S.J.; Nolan, D.; Gandy, S.E.; Martins, R.N. Apolipoprotein E, Cholesterol Metabolism, Diabetes, and the Convergence of Risk Factors for Alzheimer’s Disease and Cardiovascular Disease. Mol. Psychiatry 2006, 11, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Mattsson, N. Understanding the Cause of Sporadic Alzheimer’s Disease. Expert Rev. Neurother. 2014, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Higgins, G. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking Pathophysiological Processes in Alzheimer’s Disease: An Updated Hypothetical Model of Dynamic Biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jack, C.R.; Wiste, H.J.; Weigand, S.D.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; Mielke, M.M.; et al. Brain Injury Biomarkers Are Not Dependent on β-Amyloid in Normal Elderly. Ann. Neurol. 2013, 73, 472–480. [Google Scholar] [CrossRef]
- Chételat, G. Alzheimer Disease: Aβ-Independent Processes-Rethinking Preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef]
- Mesulam, M.-M. Cholinergic Circuitry of the Human Nucleus Basalis and Its Fate in Alzheimer’s Disease: Human Cholinergic Circuitry. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The Cholinergic System in Aging and Neuronal Degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Francis, P.T. The Interplay of Neurotransmitters in Alzheimer’s Disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef]
- Beach, T.G.; Kuo, Y.-M.; Spiegel, K.; Emmerling, M.R.; Sue, L.I.; Kokjohn, K.; Roher, A.E. The Cholinergic Deficit Coincides with Aβ Deposition at the Earliest Histopathologic Stages of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2000, 59, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, Y.; Lee, J.E. Metabolism-Centric Overview of the Pathogenesis of Alzheimer’s Disease. Yonsei Med. J. 2017, 58, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the Diagnostic Investigation of Alzheimer’s Disease and Other Dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group* under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing Research Diagnostic Criteria for Alzheimer’s Disease: The IWG-2 Criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Jack, C.R.; Holtzman, D.M. Biomarker Modeling of Alzheimer’s Disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef]
- Cummings, J. The National Institute on Aging-Alzheimer’s Association Framework on Alzheimer’s Disease: Application to Clinical Trials. Alzheimer’s Dement. 2019, 15, 172–178. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Babiloni, C.; Lopez, S.; Del Percio, C.; Noce, G.; Pascarelli, M.T.; Lizio, R.; Teipel, S.J.; González-Escamilla, G.; Bakardjian, H.; George, N.; et al. Resting-State Posterior Alpha Rhythms Are Abnormal in Subjective Memory Complaint Seniors with Preclinical Alzheimer’s Neuropathology and High Education Level: The INSIGHT-PreAD Study. Neurobiol. Aging 2020, 90, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Epelbaum, S.; Nyasse, F.; Bakardjian, H.; Gagliardi, G.; Uspenskaya, O.; Houot, M.; Lista, S.; Cacciamani, F.; Potier, M.-C.; et al. Cognitive and Neuroimaging Features and Brain β-Amyloidosis in Individuals at Risk of Alzheimer’s Disease (INSIGHT-PreAD): A Longitudinal Observational Study. Lancet Neurol. 2018, 17, 335–346. [Google Scholar] [CrossRef]
- Soldan, A.; Pettigrew, C.; Cai, Q.; Wang, J.; Wang, M.-C.; Moghekar, A.; Miller, M.I.; Albert, M.; BIOCARD Research Team. Cognitive Reserve and Long-Term Change in Cognition in Aging and Preclinical Alzheimer’s Disease. Neurobiol. Aging 2017, 60, 164–172. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, P.; Seo, S.W.; Roh, J.H.; Oh, M.; Oh, J.S.; Oh, S.J.; Kim, J.S.; Jeong, Y. Neural Substrates of Cognitive Reserve in Alzheimer’s Disease Spectrum and Normal Aging. NeuroImage 2019, 186, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Gavrilova, S.I.; Samsonova, A.; Barreto, G.E.; Aliev, G. Mild Cognitive Impairment Due to Alzheimer Disease: Contemporary Approaches to Diagnostics and Pharmacological Intervention. Pharmacol. Res. 2018, 129, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Knopman, D.S. Classification and Epidemiology of MCI. Clin. Geriatr. Med. 2013, 29, 753–772. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Xu, W.; Li, J.-Q.; Wang, J.; Lai, T.-J.; et al. The Prevalence of Neuropsychiatric Symptoms in Alzheimer’s Disease: Systematic Review and Meta-Analysis. J. Affect. Disord. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An Unbiased Descriptive Classification Scheme for Alzheimer Disease Biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum. Med. Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The Projected Effect of Risk Factor Reduction on Alzheimer’s Disease Prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer Disease: Epidemiology, Diagnostic Criteria, Risk Factors and Biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Deckers, K.; van Boxtel, M.P.J.; Schiepers, O.J.G.; de Vugt, M.; Muñoz Sánchez, J.L.; Anstey, K.J.; Brayne, C.; Dartigues, J.-F.; Engedal, K.; Kivipelto, M.; et al. Target Risk Factors for Dementia Prevention: A Systematic Review and Delphi Consensus Study on the Evidence from Observational Studies. Int. J. Geriatr. Psychiatry 2015, 30, 234–246. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.d.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.d.G. Alzheimer’s Disease: Risk Factors and Potentially Protective Measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Bienias, J.L.; Aggarwal, N.T.; Wilson, R.S.; Bennett, D.A.; Shah, R.C.; Evans, D.A. Change in Risk of Alzheimer Disease over Time. Neurology 2010, 75, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.J.; Guerchet, M.M.; Prina, M. The Epidemiology and Impact of Dementia: Current State and Future Trends. WHO Thematic Briefing. 2015. Available online: https://hal.archives-ouvertes.fr/hal-03517019 (accessed on 4 June 2022).
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic Meta-Analyses of Alzheimer Disease Genetic Association Studies: The AlzGene Database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B. Genetics of Dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.-T.; Wang, H.-F.; Han, P.-R.; Tan, C.-C.; Wang, C.; Meng, X.-F.; Risacher, S.L.; Saykin, A.J.; Tan, L. APOE Genotype and Neuroimaging Markers of Alzheimer’s Disease: Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual Dimorphism in Predisposition to Alzheimer’s Disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Nebel, R.A.; Aggarwal, N.T.; Barnes, L.L.; Gallagher, A.; Goldstein, J.M.; Kantarci, K.; Mallampalli, M.P.; Mormino, E.C.; Scott, L.; Yu, W.H.; et al. Understanding the Impact of Sex and Gender in Alzheimer’s Disease: A Call to Action. Alzheimer’s Dement. 2018, 14, 1171–1183. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and Sex: Triad of Risk of Alzheimer’s Disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Sidney, S.; Selby, J.; Johnston, S.C.; Yaffe, K. Midlife Cardiovascular Risk Factors and Risk of Dementia in Late Life. Neurology 2005, 64, 277–281. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Yu, J.-T.; Xu, W.; Tan, C.-C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-Based Prevention of Alzheimer’s Disease: Systematic Review and Meta-Analysis of 243 Observational Prospective Studies and 153 Randomised Controlled Trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef]
- Edwards, G.A., III; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef]
- Uauy, R.; Dangour, A.D. Nutrition in Brain Development and Aging: Role of Essential Fatty Acids. Nutr. Rev. 2006, 64, S24–S33, discussion S72–S91. [Google Scholar] [CrossRef]
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Korade, Z.; Kenworthy, A.K. Lipid Rafts, Cholesterol, and the Brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef]
- Luchsinger, J.A. Adiposity, Hyperinsulinemia, Diabetes and Alzheimer’s Disease. Eur. J. Pharmacol. 2008, 585, 119–129. [Google Scholar] [CrossRef]
- Hildreth, K.L.; Pelt, R.E.; Schwartz, R.S. Obesity, Insulin Resistance, and Alzheimer’s Disease. Obesity 2012, 20, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s Disease a Type 3 Diabetes? A Critical Appraisal. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Thomas, J.; Thomas, C.J.; Radcliffe, J.; Itsiopoulos, C. Omega-3 Fatty Acids in Early Prevention of Inflammatory Neurodegenerative Disease: A Focus on Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.-B.; Zhang, Z.; Luo, P.; Wang, S.-S.; Peng, Y.; Chu, S.-F.; Chen, N.-H. Lipid Metabolism in Alzheimer’s Disease. Brain Res. Bull. 2019, 144, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of Lipids in Alzheimer’s Disease and Their Role as Potential Biomarkers. Alzheimer’s Dement. 2017, 13, 810–827. [Google Scholar] [CrossRef] [PubMed]
- Justice, N.J. The Relationship between Stress and Alzheimer’s Disease. Neurobiol. Stress 2018, 8, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, M.; Du, Y.; Dodge, H.H.; Ratcliff, G.G.; Chang, C.-C.H. Depressive Symptoms and Cognitive Decline in Late Life: A Prospective Epidemiological Study. Arch. Gen. Psychiatry 2006, 63, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head Injury as a Risk Factor for Alzheimer’s Disease: The Evidence 10 Years on; a Partial Replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.; Ee, N.; Peters, J.; Booth, A.; Mudway, I.; Anstey, K.J. Air Pollution and Dementia: A Systematic Review. J. Alzheimer’s Dis. 2019, 70, S145–S163. [Google Scholar] [CrossRef]
- Cenini, G.; Hebisch, M.; Iefremova, V.; Flitsch, L.J.; Breitkreuz, Y.; Tanzi, R.E.; Kim, D.Y.; Peitz, M.; Brüstle, O. Dissecting Alzheimer’s Disease Pathogenesis in Human 2D and 3D Models. Mol. Cell. Neurosci. 2020, 110, 103568. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Jodat, Y.A.; Samanipour, R.; Zorzi, G.; Zhu, K.; Hirano, M.; Chang, K.; Arnaout, A.; Hassan, S.; Matharu, N.; et al. Toward a Neurospheroid Niche Model: Optimizing Embedded 3D Bioprinting for Fabrication of Neurospheroid Brain-like Co-Culture Constructs. Biofabrication 2020, 13, 015014. [Google Scholar] [CrossRef] [PubMed]
- Morsink, M.A.J.; Willemen, N.G.A.; Leijten, J.; Bansal, R.; Shin, S.R. Immune Organs and Immune Cells on a Chip: An Overview of Biomedical Applications. Micromachines 2020, 11, 849. [Google Scholar] [CrossRef]
- Boni, R.; Ali, A.; Shavandi, A.; Clarkson, A.N. Current and Novel Polymeric Biomaterials for Neural Tissue Engineering. J. Biomed. Sci. 2018, 25, 90. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D Human Induced Pluripotent Stem Cell-Derived Cultures for Neurodegenerative Disease Modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; Macleod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of Treatment Effects between Animal Experiments and Clinical Trials: Systematic Review. BMJ 2007, 334, 197. [Google Scholar] [CrossRef]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stüber, K.; Esselmann, H.; Wiltfang, J.; Brüstle, O.; Walter, J. Presenilin-1 L166P Mutant Human Pluripotent Stem Cell–Derived Neurons Exhibit Partial Loss of γ-Secretase Activity in Endogenous Amyloid-β Generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Stüber, K.; Wunderlich, P.; Ladewig, J.; Kesavan, J.C.; Vandenberghe, R.; Vandenbulcke, M.; van Damme, P.; Walter, J.; Brüstle, O.; et al. APP Processing in Human Pluripotent Stem Cell-Derived Neurons Is Resistant to NSAID-Based γ-Secretase Modulation. Stem Cell Rep. 2013, 1, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-C.; Muratore, C.R.; Gierahn, T.M.; Sullivan, S.E.; Srikanth, P.; Jager, P.L.D.; Love, J.C.; Young-Pearse, T.L. Single-Cell Detection of Secreted Aβ and SAPPα from Human IPSC-Derived Neurons and Astrocytes. J. Neurosci. 2016, 36, 1730–1746. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Olivé, M.G.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant IPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant IPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and IPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef]
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β Oligomers in Cellular Models of Alzheimer’s Disease. J. Neurochem. 2020, 155, 348–369. [Google Scholar] [CrossRef]
- Hernández-Sapiéns, M.A.; Reza-Zaldívar, E.E.; Cevallos, R.R.; Márquez-Aguirre, A.L.; Gazarian, K.; Canales-Aguirre, A.A. A Three-Dimensional Alzheimer’s Disease Cell Culture Model Using IPSC-Derived Neurons Carrying A246E Mutation in PSEN1. Front. Cell. Neurosci. 2020, 14, 151. [Google Scholar] [CrossRef]
- Ranjan, V.D.; Qiu, L.; Lee, J.W.-L.; Chen, X.; Jang, S.E.; Chai, C.; Lim, K.-L.; Tan, E.-K.; Zhang, Y.; Huang, W.M.; et al. A Microfiber Scaffold-Based 3D in Vitro Human Neuronal Culture Model of Alzheimer’s Disease. Biomater. Sci. 2020, 8, 4861–4874. [Google Scholar] [CrossRef]
- Papadimitriou, C.; Celikkaya, H.; Cosacak, M.I.; Mashkaryan, V.; Bray, L.; Bhattarai, P.; Brandt, K.; Hollak, H.; Chen, X.; He, S.; et al. 3D Culture Method for Alzheimer’s Disease Modeling Reveals Interleukin-4 Rescues Aβ42-Induced Loss of Human Neural Stem Cell Plasticity. Dev. Cell 2018, 46, 85–101.e8. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D Human Brain–like Tissue Model of Herpes-Induced Alzheimer’s Disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Sanchez, C.V.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-Β42/40 Ratio Drives Tau Pathology in 3D Human Neural Cell Culture Models of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Ao, Z.; Hu, L.; Moon, Y.; Wu, Z.; Lu, H.-C.; Kim, J.; Guo, F. Acoustofluidic Assembly of 3D Neurospheroids to Model Alzheimer’s Disease. Analyst 2020, 145, 6243–6253. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Chen, G.; Chen, K.S.; Knox, J.; Inglis, J.; Bernard, A.; Martin, S.J.; Justice, A.; McConlogue, L.; Games, D.; Freedman, S.B.; et al. A Learning Deficit Related to Age and β-Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef]
- Ochiishi, T.; Kaku, M.; Kiyosue, K.; Doi, M.; Urabe, T.; Hattori, N.; Shimura, H.; Ebihara, T. New Alzheimer’s Disease Model Mouse Specialized for Analyzing the Function and Toxicity of Intraneuronal Amyloid β Oligomers. Sci. Rep. 2019, 9, 17368. [Google Scholar] [CrossRef]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E.; et al. Intracerebral Injection of Preformed Synthetic Tau Fibrils Initiates Widespread Tauopathy and Neuronal Loss in the Brains of Tau Transgenic Mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Banks, R.A.; Kim, B.; Xu, H.; Changolkar, L.; Leight, S.N.; Riddle, D.M.; Li, C.; Gathagan, R.J.; Brown, H.J.; et al. GFP-Mutant Human Tau Transgenic Mice Develop Tauopathy Following CNS Injections of Alzheimer’s Brain-Derived Pathological Tau or Synthetic Mutant Human Tau Fibrils. J. Neurosci. 2017, 37, 11485–11494. [Google Scholar] [CrossRef]
- Faucher, P.; Mons, N.; Micheau, J.; Louis, C.; Beracochea, D.J. Hippocampal Injections of Oligomeric Amyloid β-Peptide (1–42) Induce Selective Working Memory Deficits and Long-Lasting Alterations of ERK Signaling Pathway. Front. Aging Neurosci. 2016, 7, 245. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of Function of the Mitochondrial Peptidase PITRM1 Induces Proteotoxic Stress and Alzheimer’s Disease-like Pathology in Human Cerebral Organoids. Mol. Psychiatry 2020, 10, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- D’Avanzo, C.; Aronson, J.; Kim, Y.H.; Choi, S.H.; Tanzi, R.E.; Kim, D.Y. Alzheimer’s in 3D Culture: Challenges and Perspectives. Bioessays 2015, 37, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Quinti, L.; Tanzi, R.E.; Kim, D.Y. 3D Culture Models of Alzheimer’s Disease: A Road Map to a “Cure-in-a-Dish”. Mol. Neurodegener. 2016, 11, 75. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, S.H.; D’Avanzo, C.; Hebisch, M.; Sliwinski, C.; Bylykbashi, E.; Washicosky, K.J.; Klee, J.B.; Brüstle, O.; Tanzi, R.E.; et al. A 3D Human Neural Cell Culture System for Modeling Alzheimer’s Disease. Nat. Protoc. 2015, 10, 985–1006. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical Considerations for Choosing a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal Models of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef]
- Clarke, J.R.; Lyra e Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazão, R.; et al. Alzheimer-associated Aβ Oligomers Impact the Central Nervous System to Induce Peripheral Metabolic Deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Latimer, C.S.; Shively, C.A.; Keene, C.D.; Jorgensen, M.J.; Andrews, R.N.; Register, T.C.; Montine, T.J.; Wilson, A.M.; Neth, B.J.; Mintz, A.; et al. A Nonhuman Primate Model of Early Alzheimer’s Disease Pathologic Change: Implications for Disease Pathogenesis. Alzheimer’s Dement. 2019, 15, 93–105. [Google Scholar] [CrossRef]
- Frye, B.M.; Craft, S.; Register, T.C.; Kim, J.; Whitlow, C.T.; Barcus, R.A.; Lockhart, S.N.; Sai, K.K.S.; Shively, C.A. Early Alzheimer’s Disease-like Reductions in Gray Matter and Cognitive Function with Aging in Nonhuman Primates. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12284. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug Treatments in Alzheimer’s Disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. Current and Future Treatments in Alzheimer’s Disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Ritter, A.; Zhong, K. Clinical Trials for Disease-Modifying Therapies in Alzheimer’s Disease: A Primer, Lessons Learned, and a Blueprint for the Future1. J. Alzheimer’s Dis. 2018, 64, S3–S22. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Chakraborty, A.; de Wit, N.M.; van der Flier, W.M.; de Vries, H.E. The Blood Brain Barrier in Alzheimer’s Disease. Vascul. Pharmacol. 2017, 89, 12–18. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood–Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Banks, W.A. Drug Delivery to the Brain in Alzheimer’s Disease: Consideration of the Blood-Brain Barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar] [CrossRef]
- Colin, J.; Thomas, M.H.; Gregory-Pauron, L.; Pinçon, A.; Lanhers, M.-C.; Corbier, C.; Claudepierre, T.; Yen, F.T.; Oster, T.; Malaplate-Armand, C. Maintenance of Membrane Organization in the Aging Mouse Brain as the Determining Factor for Preventing Receptor Dysfunction and for Improving Response to Anti-Alzheimer Treatments. Neurobiol. Aging 2017, 54, 84–93. [Google Scholar] [CrossRef]
- Poon, C.H.; Wang, Y.; Fung, M.-L.; Zhang, C.; Lim, L.W. Rodent Models of Amyloid-Beta Feature of Alzheimer’s Disease: Development and Potential Treatment Implications. Aging Dis. 2020, 11, 1235. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Klimova, B.; Kuca, K. Alzheimer’s Disease: Potential Preventive, Non-Invasive, Intervention Strategies in Lowering the Risk of Cognitive Decline—A Review Study. J. Appl. Biomed. 2015, 13, 257–261. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle Interventions to Prevent Cognitive Impairment, Dementia and Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Kivipelto Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World-Wide FINGERS. J. Prev. Alzheimer’s Dis. 2019, 7, 29–36. [Google Scholar] [CrossRef]
- Lehtisalo, J.; Levälahti, E.; Lindström, J.; Hänninen, T.; Paajanen, T.; Peltonen, M.; Antikainen, R.; Laatikainen, T.; Strandberg, T.; Soininen, H.; et al. Dietary Changes and Cognition over 2 Years within a Multidomain Intervention Trial-The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). Alzheimer’s Dement. 2019, 15, 410–417. [Google Scholar] [CrossRef]
- Olazarán, J.; Reisberg, B.; Clare, L.; Cruz, I.; Peña-Casanova, J.; Del Ser, T.; Woods, B.; Beck, C.; Auer, S.; Lai, C.; et al. Nonpharmacological Therapies in Alzheimer’s Disease: A Systematic Review of Efficacy. Dement. Geriatr. Cogn. Disord. 2010, 30, 161–178. [Google Scholar] [CrossRef]
- Van Wijk, N.; Broersen, L.M.; de Wilde, M.C.; Hageman, R.J.J.; Groenendijk, M.; Sijben, J.W.C.; Kamphuis, P.J.G.H. Targeting Synaptic Dysfunction in Alzheimer’s Disease by Administering a Specific Nutrient Combination. J. Alzheimer’s Dis. 2013, 38, 459–479. [Google Scholar] [CrossRef]
- Rasmussen, J. The LipiDiDiet Trial: What Does It Add to the Current Evidence for Fortasyn Connect in Early Alzheimer’s Disease? Clin. Interv. Aging 2019, 14, 1481–1492. [Google Scholar] [CrossRef]
- Singh-Manoux, A.; Kivimaki, M.; Glymour, M.M.; Elbaz, A.; Berr, C.; Ebmeier, K.P.; Ferrie, J.E.; Dugravot, A. Timing of Onset of Cognitive Decline: Results from Whitehall II Prospective Cohort Study. BMJ 2012, 344, d7622. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural Mechanisms of Ageing and Cognitive Decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef]
- Colin, J.; Gregory-Pauron, L.; Lanhers, M.-C.; Claudepierre, T.; Corbier, C.; Yen, F.T.; Malaplate-Armand, C.; Oster, T. Membrane Raft Domains and Remodeling in Aging Brain. Biochimie 2016, 130, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Raz, N.; Rodrigue, K.M. Differential Aging of the Brain: Patterns, Cognitive Correlates and Modifiers. Neurosci. Biobehav. Rev. 2006, 30, 730–748. [Google Scholar] [CrossRef]
- Youdim, K.A.; Martin, A.; Joseph, J.A. Essential Fatty Acids and the Brain: Possible Health Implications. Int. J. Dev. Neurosci. 2000, 18, 383–399. [Google Scholar] [CrossRef]
- Latifi, S.; Tamayol, A.; Habibey, R.; Sabzevari, R.; Kahn, C.; Geny, D.; Eftekharpour, E.; Annabi, N.; Blau, A.; Linder, M.; et al. Natural Lecithin Promotes Neural Network Complexity and Activity. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Latifi, S.; Kahn, C.J.F.; Tamayol, A.; Habibey, R.; Passeri, E.; Linder, M.; Arab-Tehrany, E. The Positive Role of Curcumin-Loaded Salmon Nanoliposomes on the Culture of Primary Cortical Neurons. Mar. Drugs 2018, 16, 218. [Google Scholar] [CrossRef] [PubMed]
- Malaplate, C.; Poerio, A.; Huguet, M.; Soligot, C.; Passeri, E.; Kahn, C.J.F.; Linder, M.; Arab-Tehrany, E.; Yen, F.T. Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons. Mar. Drugs 2019, 17, 406. [Google Scholar] [CrossRef]
- Salem, N.; Litman, B.; Kim, H.Y.; Gawrisch, K. Mechanisms of Action of Docosahexaenoic Acid in the Nervous System. Lipids 2001, 36, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty Acid Composition of Brain Phospholipids in Aging and in Alzheimer’s Disease. Lipids 1991, 26, 421–425. [Google Scholar] [CrossRef]
- Weiser, M.J.; Butt, C.M.; Mohajeri, M.H. Docosahexaenoic Acid and Cognition throughout the Lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef]
- Bazan, N.G.; Molina, M.F.; Gordon, W.C. Docosahexaenoic Acid Signalolipidomics in Nutrition: Significance in Aging, Neuroinflammation, Macular Degeneration, Alzheimer’s, and Other Neurodegenerative Diseases. Annu. Rev. Nutr. 2011, 31, 321–351. [Google Scholar] [CrossRef]
- Daiello, L.A.; Gongvatana, A.; Dunsiger, S.; Cohen, R.A.; Ott, B.R. Alzheimer’s Disease Neuroimaging Initiative Association of Fish Oil Supplement Use with Preservation of Brain Volume and Cognitive Function. Alzheimer’s Dement. 2015, 11, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagl, S.; Hener, U.; Müller, W.E.; Kögel, D. Liposome-Incorporated DHA Increases Neuronal Survival by Enhancing Non-Amyloidogenic APP Processing. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Oster, T.; Pillot, T. Docosahexaenoic Acid and Synaptic Protection in Alzheimer’s Disease Mice. Biochim. Biophys. Acta 2010, 1801, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish Consumption, Long-Chain Omega-3 Fatty Acids and Risk of Cognitive Decline or Alzheimer Disease: A Complex Association. Nat. Clin. Pract. Neurol. 2009, 5, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the Blood-Brain Barrier with Nanoparticles. J. Control. Release 2018, 270, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The Extracellular Matrix of the Blood–Brain Barrier: Structural and Functional Roles in Health, Aging, and Alzheimer’s Disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef]
- Grabrucker, A.M.; Chhabra, R.; Belletti, D.; Forni, F.; Vandelli, M.A.; Ruozi, B.; Tosi, G. Nanoparticles as Blood–Brain Barrier Permeable CNS Targeted Drug Delivery Systems. In The Blood Brain Barrier (BBB); Fricker, G., Ott, M., Mahringer, A., Eds.; Topics in Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2013; Volume 10, pp. 71–89. ISBN 978-3-662-43786-5. [Google Scholar]
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.-F. Brain Barrier Systems: A New Frontier in Metal Neurotoxicological Research. Toxicol. Appl. Pharmacol. 2003, 192, 1–11. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horne, R.W. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Li, J.; Elkhoury, K.; Barbieux, C.; Linder, M.; Grandemange, S.; Tamayol, A.; Francius, G.; Arab-Tehrany, E. Effects of Bioactive Marine-Derived Liposomes on Two Human Breast Cancer Cell Lines. Mar. Drugs 2020, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Novel Applications of Liposomes. Trends Biotechnol. 1998, 16, 307–321. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Marčelja, S.; Horn, R.G. Physical Principles of Membrane Organization. Quart. Rev. Biophys. 1980, 13, 121–200. [Google Scholar] [CrossRef]
- Hasan, M.; Elkhoury, K.; Belhaj, N.; Kahn, C.; Tamayol, A.; Barberi-Heyob, M.; Arab-Tehrany, E.; Linder, M. Growth-Inhibitory Effect of Chitosan-Coated Liposomes Encapsulating Curcumin on MCF-7 Breast Cancer Cells. Mar. Drugs 2020, 18, 217. [Google Scholar] [CrossRef] [PubMed]
- Elkhoury, K.; Russell, C.S.; Sanchez-Gonzalez, L.; Mostafavi, A.; Williams, T.J.; Kahn, C.; Peppas, N.A.; Arab-Tehrany, E.; Tamayol, A. Soft-Nanoparticle Functionalization of Natural Hydrogels for Tissue Engineering Applications. Adv. Healthcare Mater. 2019, 8, 1900506. [Google Scholar] [CrossRef]
- Hasan, M.; Elkhoury, K.; Kahn, C.J.F.; Arab-Tehrany, E.; Linder, M. Preparation, Characterization, and Release Kinetics of Chitosan-Coated Nanoliposomes Encapsulating Curcumin in Simulated Environments. Molecules 2019, 24, 2023. [Google Scholar] [CrossRef]
- Elkhoury, K.; Koçak, P.; Kang, A.; Arab-Tehrany, E.; Ellis Ward, J.; Shin, S.R. Engineering Smart Targeting Nanovesicles and Their Combination with Hydrogels for Controlled Drug Delivery. Pharmaceutics 2020, 12, 849. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting Liposomes for Oral Drug Delivery. Acta Pharm. Sin. B 2019, 9, 36–48. [Google Scholar] [CrossRef]
- Bianchi, A.; Velot, É.; Kempf, H.; Elkhoury, K.; Sanchez-Gonzalez, L.; Linder, M.; Kahn, C.; Arab-Tehrany, E. Nanoliposomes from Agro-Resources as Promising Delivery Systems for Chondrocytes. Int. J. Mol. Sci. 2020, 21, 3436. [Google Scholar] [CrossRef]
- Spuch, C.; Navarro, C. Liposomes for Targeted Delivery of Active Agents against Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). J. Drug Deliv. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Chiang, C.-F.; Chen, L.-F.; Liang, P.-C.; Hsieh, W.-Y.; Lin, W.-L. Polymersomes Conjugated with Des-Octanoyl Ghrelin and Folate as a BBB-Penetrating Cancer Cell-Targeting Delivery System. Biomaterials 2014, 35, 4066–4081. [Google Scholar] [CrossRef] [PubMed]
- Mourtas, S.; Lazar, A.N.; Markoutsa, E.; Duyckaerts, C.; Antimisiaris, S.G. Multifunctional Nanoliposomes with Curcumin–Lipid Derivative and Brain Targeting Functionality with Potential Applications for Alzheimer Disease. Eur. J. Med. Chem. 2014, 80, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Markoutsa, E.; Papadia, K.; Giannou, A.D.; Spella, M.; Cagnotto, A.; Salmona, M.; Stathopoulos, G.T.; Antimisiaris, S.G. Mono and Dually Decorated Nanoliposomes for Brain Targeting, In Vitro and In Vivo Studies. Pharm. Res. 2014, 31, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Markoutsa, E.; Papadia, K.; Clemente, C.; Flores, O.; Antimisiaris, S.G. Anti-Aβ-MAb and Dually Decorated Nanoliposomes: Effect of Aβ1-42 Peptides on Interaction with HCMEC/D3 Cells. Eur. J. Pharm. Biopharm. 2012, 81, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, L.; Qin, Y.; Yin, Y.; Tang, J.; Tang, W.; Sun, X.; Zhang, Z.; Liu, J.; He, Q. Lactoferrin-Modified Procationic Liposomes as a Novel Drug Carrier for Brain Delivery. Eur. J. Pharm. Sci. 2010, 40, 94–102. [Google Scholar] [CrossRef]
- Chen, H.; Qin, Y.; Zhang, Q.; Jiang, W.; Tang, L.; Liu, J.; He, Q. Lactoferrin Modified Doxorubicin-Loaded Procationic Liposomes for the Treatment of Gliomas. Eur. J. Pharm. Sci. 2011, 44, 164–173. [Google Scholar] [CrossRef]
- Joshi, S.; Singh-Moon, R.P.; Ellis, J.A.; Chaudhuri, D.B.; Wang, M.; Reif, R.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Cerebral Hypoperfusion-Assisted Intra-Arterial Deposition of Liposomes in Normal and Glioma-Bearing Rats. Neurosurgery 2015, 76, 92–100. [Google Scholar] [CrossRef]
- Joshi, S.; Singh-Moon, R.P.; Wang, M.; Chaudhuri, D.B.; Holcomb, M.; Straubinger, N.L.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Transient Cerebral Hypoperfusion Assisted Intraarterial Cationic Liposome Delivery to Brain Tissue. J. Neurooncol. 2014, 118, 73–82. [Google Scholar] [CrossRef]
- Joshi, S.; Singh-Moon, R.; Wang, M.; Chaudhuri, D.B.; Ellis, J.A.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Cationic Surface Charge Enhances Early Regional Deposition of Liposomes after Intracarotid Injection. J. Neurooncol. 2014, 120, 489–497. [Google Scholar] [CrossRef]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-Targeted Liposome Design: Challenges and Fundamental Considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Chang, N.; Sun, S.; Li, M.; Yu, H.; Liu, M.; Liu, X.; Wang, G.; Li, H.; Liu, X.; et al. The Role of Glucose Transporters in the Distribution of P-Aminophenyl-α-d-Mannopyranoside Modified Liposomes within Mice Brain. J. Control. Release 2014, 182, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Li, X.; Guan, M.; Li, X.; Hai, L.; Wu, Y. Design, Synthesis and Biological Evaluation of Multivalent Glucosides with High Affinity as Ligands for Brain Targeting Liposomes. Eur. J. Med. Chem. 2014, 72, 110–118. [Google Scholar] [CrossRef]
- Lindqvist, A.; Rip, J.; van Kregten, J.; Gaillard, P.J.; Hammarlund-Udenaes, M. In Vivo Functional Evaluation of Increased Brain Delivery of the Opioid Peptide DAMGO by Glutathione-PEGylated Liposomes. Pharm. Res. 2016, 33, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Rip, J.; van Kregten, J.; van den Heuvel, A.; van der Pol, S.; van der Boom, B.; Reijerkerk, A.; Chen, L.; de Boer, M.; Gaillard, P.; et al. Glutathione Conjugation Dose-Dependently Increases Brain-Specific Liposomal Drug Delivery in Vitro and in Vivo. Drug Discov. Today Technol. 2016, 20, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-X.; Zhao, W.-Y.; Liu, L.; Ju, R.-J.; Mu, L.-M.; Zhao, Y.; Zeng, F.; Xie, H.-J.; Yan, Y.; Lu, W.-L. A Nanostructure of Functional Targeting Epirubicin Liposomes Dually Modified with Aminophenyl Glucose and Cyclic Pentapeptide Used for Brain Glioblastoma Treatment. Oncotarget 2015, 6, 32681–32700. [Google Scholar] [CrossRef]
- Li, X.; Tang, W.; Jiang, Y.; Wang, X.; Wang, Y.; Cheng, L.; Meng, X. Multifunctional Targeting Vinorelbine plus Tetrandrine Liposomes for Treating Brain Glioma along with Eliminating Glioma Stem Cells. Oncotarget 2016, 7, 24604–24622. [Google Scholar] [CrossRef]
- Liu, Y.; Ran, R.; Chen, J.; Kuang, Q.; Tang, J.; Mei, L.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Paclitaxel Loaded Liposomes Decorated with a Multifunctional Tandem Peptide for Glioma Targeting. Biomaterials 2014, 35, 4835–4847. [Google Scholar] [CrossRef]
- Markoutsa, E.; Mourtas, S.; Bereczki, E.; Zona, C.; Ferla, B.; Nicotra, F.; Flores, O.; Pei, J.-J.; Antimisiaris, S. Comparison of Various Types of Ligand Decorated Nanoliposomes for Their Ability to Inhibit Amyloid Aggregation and to Reverse Amyloid Cytotoxicity. Curr. Top. Med. Chem. 2015, 15, 2267–2276. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H.; Huang, Q.; Peng, C.; Yao, L.; Chen, H.; Qiu, Z.; Wu, Y.; Wang, L.; Chen, W. Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation. Theranostics 2019, 9, 1714–1727. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular Membrane Vesicles as a Mechanism of Cell-to-Cell Communication: Advantages and Disadvantages. Am. J. Physiol.-Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Su, C. Design Strategies and Application Progress of Therapeutic Exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Antimisiaris, S.; Mourtas, S.; Marazioti, A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A Comprehensive Overview of Exosomes as Drug Delivery Vehicles—Endogenous Nanocarriers for Targeted Cancer Therapy. Biochim. Biophys. Acta-Rev. Cancer 2014, 1846, 75–87. [Google Scholar] [CrossRef]
- Bang, C.; Thum, T. Exosomes: New Players in Cell–Cell Communication. Int. J. Biochem. Cell Biol. 2012, 44, 2060–2064. [Google Scholar] [CrossRef]
- Fais, S.; O’Driscoll, L.; Borras, F.E.; Buzas, E.; Camussi, G.; Cappello, F.; Carvalho, J.; Cordeiro da Silva, A.; Del Portillo, H.; El Andaloussi, S.; et al. Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano 2016, 10, 3886–3899. [Google Scholar] [CrossRef]
- Yakimchuk, K. Exosomes: Isolation Methods and Specific Markers. Mater. Methods 2015, 5, 1450. [Google Scholar] [CrossRef]
- Saeedi, S.; Israel, S.; Nagy, C.; Turecki, G. The Emerging Role of Exosomes in Mental Disorders. Transl. Psychiatry 2019, 9, 122. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-Inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Molina, C.; Sandoval, M.; Henzi, R.; Ramírez, J.P.; Varas-Godoy, M.; Luarte, A.; Lafourcade, C.A.; Lopez-Verrilli, A.; Smalla, K.-H.; Kaehne, T.; et al. Small Extracellular Vesicles in Rat Serum Contain Astrocyte-Derived Protein Biomarkers of Repetitive Stress. Int. J. Neuropsychopharmacol. 2019, 22, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as Drug Delivery Vehicles for Parkinson’s Disease Therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, D.; Liu, Z.; Zhou, Y.; Chu, D.; Li, X.; Jiang, X.; Hou, D.; Chen, X.; Chen, Y.; et al. Targeted Exosome-Mediated Delivery of Opioid Receptor Mu SiRNA for the Treatment of Morphine Relapse. Sci. Rep. 2015, 5, 17543. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.E.; Scioli Montoto, S. Routes of Drug Administration. In ADME Processes in Pharmaceutical Sciences; Talevi, A., Quiroga, P.A.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 97–133. ISBN 978-3-319-99592-2. [Google Scholar]
- Barnabas, W. Drug Targeting Strategies into the Brain for Treating Neurological Diseases. J. Neurosci. Methods 2019, 311, 133–146. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Bannerjee, S.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug Delivery Systems: An Updated Review. Int. J. Pharma. Investig. 2012, 2, 2. [Google Scholar] [CrossRef]
- Rhea, E.M.; Salameh, T.S.; Banks, W.A. Routes for the Delivery of Insulin to the Central Nervous System: A Comparative Review. Exp. Neurol. 2019, 313, 10–15. [Google Scholar] [CrossRef]
- Begley, D.J. Delivery of Therapeutic Agents to the Central Nervous System: The Problems and the Possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef]
- Chapman, C.D.; Frey, W.H.; Craft, S.; Danielyan, L.; Hallschmid, M.; Schiöth, H.B.; Benedict, C. Intranasal Treatment of Central Nervous System Dysfunction in Humans. Pharm. Res. 2013, 30, 2475–2484. [Google Scholar] [CrossRef]
- Mittal, D.; Ali, A.; Md, S.; Baboota, S.; Sahni, J.K.; Ali, J. Insights into Direct Nose to Brain Delivery: Current Status and Future Perspective. Drug Deliv. 2014, 21, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal Drug Delivery: How, Why and What For? J. Pharm. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in Nose-to-Brain Drug Delivery: A Review of Non-Clinical Brain Targeting Studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Alonso, M.J. Nose-to-Brain Peptide Delivery—The Potential of Nanotechnology. Bioorg. Med. Chem. 2018, 26, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal Delivery to the Central Nervous System: Mechanisms and Experimental Considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The Nasal Approach to Delivering Treatment for Brain Diseases: An Anatomic, Physiologic, and Delivery Technology Overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef]
- Guennoun, R.; Fréchou, M.; Gaignard, P.; Liere, P.; Slama, A.; Schumacher, M.; Denier, C.; Mattern, C. Intranasal Administration of Progesterone: A Potential Efficient Route of Delivery for Cerebroprotection after Acute Brain Injuries. Neuropharmacology 2019, 145, 283–291. [Google Scholar] [CrossRef]
- Guastella, A.J.; Einfeld, S.L.; Gray, K.M.; Rinehart, N.J.; Tonge, B.J.; Lambert, T.J.; Hickie, I.B. Intranasal Oxytocin Improves Emotion Recognition for Youth with Autism Spectrum Disorders. Biol. Psychiatry 2010, 67, 692–694. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal Delivery of Biologics to the Central Nervous System. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Lin, Q.; Mao, K.-L.; Tian, F.-R.; Yang, J.-J.; Chen, P.-P.; Xu, J.; Fan, Z.-L.; Zhao, Y.-P.; Li, W.-F.; Zheng, L.; et al. Brain Tumor-Targeted Delivery and Therapy by Focused Ultrasound Introduced Doxorubicin-Loaded Cationic Liposomes. Cancer Chemother. Pharmacol. 2016, 77, 269–280. [Google Scholar] [CrossRef]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical Trial of Blood-Brain Barrier Disruption by Pulsed Ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Dubey, S.; Yeung, S.; Hough, O.; Eterman, N.; Aubert, I.; Hynynen, K. Alzheimer Disease in a Mouse Model: MR Imaging-Guided Focused Ultrasound Targeted to the Hippocampus Opens the Blood-Brain Barrier and Improves Pathologic Abnormalities and Behavior. Radiology 2014, 273, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-H.; Liu, H.-L.; Ting, C.-Y.; Lee, Y.-H.; Huang, C.-Y.; Ma, Y.-J.; Wei, K.-C.; Yen, T.-C.; Yeh, C.-K. Submicron-Bubble-Enhanced Focused Ultrasound for Blood–Brain Barrier Disruption and Improved CNS Drug Delivery. PLoS ONE 2014, 9, e96327. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood-Brain Barrier Opening in Alzheimer’s Disease Using MR-Guided Focused Ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Jayant, R.D.; Sagar, V.; Nair, M. The Potential of Magneto-Electric Nanocarriers for Drug Delivery. Expert Opin. Drug Deliv. 2014, 11, 1635–1646. [Google Scholar] [CrossRef]
- Samandari, M.; Aghabaglou, F.; Nuutila, K.; Derakhshandeh, H.; Zhang, Y.; Endo, Y.; Harris, S.; Barnum, L.; Kreikemeier-Bower, C.; Arab-Tehrany, E.; et al. Miniaturized Needle Array-Mediated Drug Delivery Accelerates Wound Healing. Adv. Healthc. Mater. 2021, 10, 2001800. [Google Scholar] [CrossRef]
- Derakhshandeh, H.; Aghabaglou, F.; McCarthy, A.; Mostafavi, A.; Wiseman, C.; Bonick, Z.; Ghanavati, I.; Harris, S.; Kreikemeier-Bower, C.; Moosavi Basri, S.M.; et al. A Wirelessly Controlled Smart Bandage with 3D-Printed Miniaturized Needle Arrays. Adv. Funct. Mater. 2020, 30, 1905544. [Google Scholar] [CrossRef]
- Barnum, L.; Quint, J.; Derakhshandeh, H.; Samandari, M.; Aghabaglou, F.; Farzin, A.; Abbasi, L.; Bencherif, S.; Memic, A.; Mostafalu, P.; et al. 3D-Printed Hydrogel-Filled Microneedle Arrays. Adv. Healthc. Mater. 2021, 10, 2001922. [Google Scholar] [CrossRef]
- Aich, K.; Singh, T.; Dang, S. Advances in Microneedle-Based Transdermal Delivery for Drugs and Peptides. Drug Deliv. Transl. Res. 2022, 12, 1556–1568. [Google Scholar] [CrossRef]
- Yan, Q.; Wang, W.; Weng, J.; Zhang, Z.; Yin, L.; Yang, Q.; Guo, F.; Wang, X.; Chen, F.; Yang, G. Dissolving Microneedles for Transdermal Delivery of Huperzine A for the Treatment of Alzheimer’s Disease. Drug Deliv. 2020, 27, 1147–1155. [Google Scholar] [CrossRef]
- Bandiwadekar, A.; Jose, J.; Khayatkashani, M.; Habtemariam, S.; Khayat Kashani, H.R.; Nabavi, S.M. Emerging Novel Approaches for the Enhanced Delivery of Natural Products for the Management of Neurodegenerative Diseases. J. Mol. Neurosci. 2022, 72, 653–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kantor, B.; Chiba-Falek, O. APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s. Int. J. Mol. Sci. 2021, 22, 1244. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s Disease: Targeting β-Amyloid and Beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.K. Anti-Prion Systems in Yeast and Inositol Polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Gorkovskiy, A.; Bezsonov, E.E.; Stroobant, E.E. Yeast and Fungal Prions. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2016; Volume 93, pp. 191–236. ISBN 978-0-12-804801-6. [Google Scholar]
- Mcdonald, J.; Dhakal, S.; Macreadie, I. Yeast Contributions to Alzheimer’s Disease. J. Human Clin. Genet. 2020, 2, 1114. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
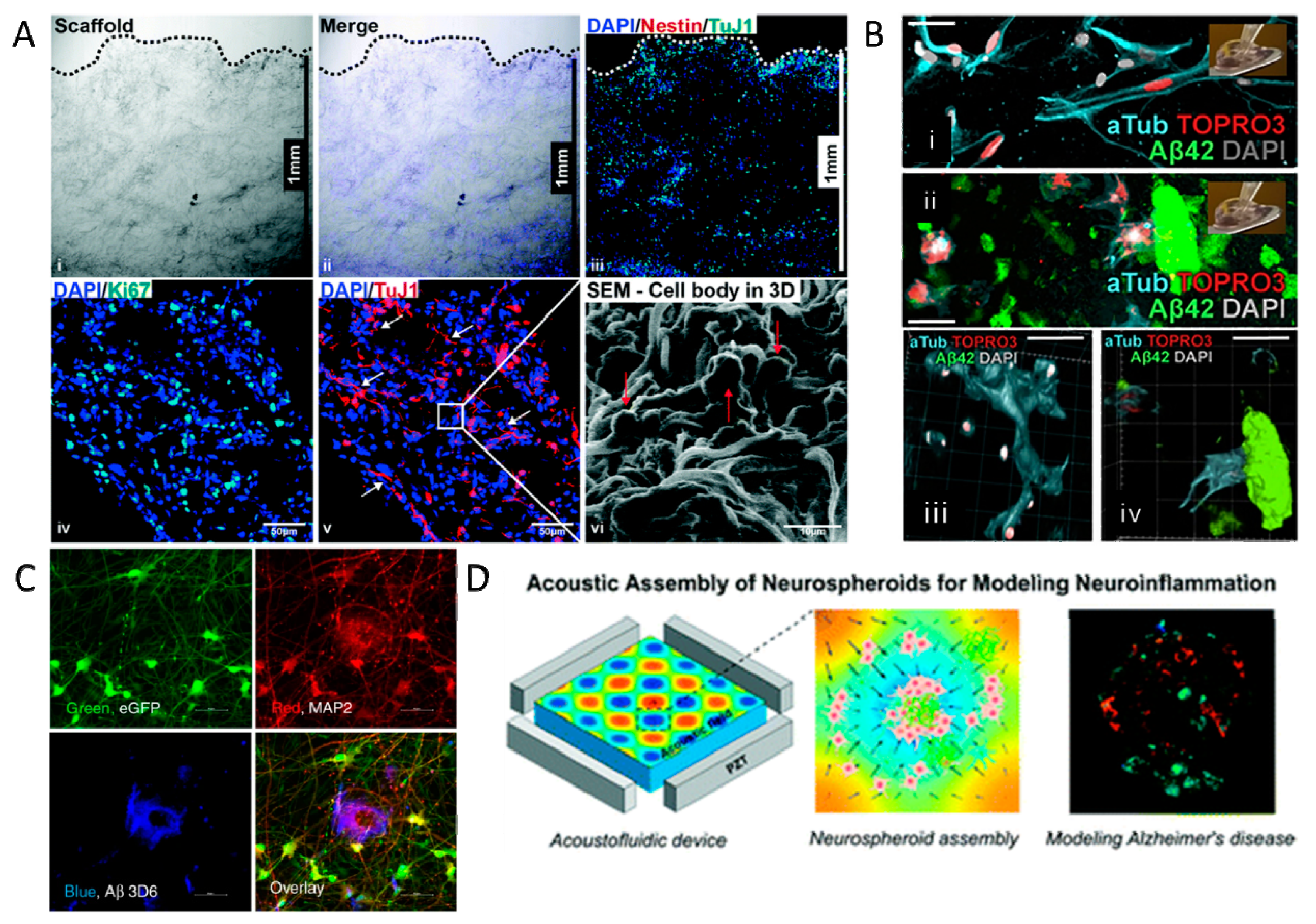
| Type | Model | Key Findings | Ref. |
|---|---|---|---|
| 2D in vitro models | hESC-derived neurons overexpressing PSEN1 | Increased Aβ42/40 ratio due to depletion of Aβ40 | [125] |
| APP K724N mutated neurons from AD patients | Increased Aβ42/40 ratio due to depletion of Aβ40 and increased secretion of Aβ42 | [126] | |
| hiPSC-derived astrocytes | Increased Aβ42/40 ratio in astrocytes is an important regulator of AD | [127] | |
| PSEN1 ΔE9 mutated hiPSC-derived astrocytes | Increased Aβ42/40 ratio, ROS, increased cytokine release | [128] | |
| PSEN1 M146L mutated hiPSC-derived astrocytes | Disturbed expression of astrocyte markers | [129] | |
| hiPSCs-derived neurons with PSEN1 A246E mutation | Defective mitochondria have a key role in AD | [130] | |
| ReN immortalized stem cell line | Mutations in APP gene show accumulation of Aβ and phosphorylated tau | [131] | |
| PC12 immortalized cell line | GLP-1 neuroprotection and findings of Aβ toxicity | [132] | |
| 3D in vitro models | PSEN1 A246E iPSC-derived neurons | Aβ aggregation without synthetic Aβ exposure or mutation induction | [133] |
| iPSC-derived NPCs encapsulated in wet electrospun PLGA | Enhanced expression of Aβ42 and p-Tau | [134] | |
| NSCs encapsulated in starPEG-heparin-based hydrogels | Increased Aβ42 causes loss of neuroplasticity. System could allow for identification of therapeutic targets | [135] | |
| Induced NSCs in silk protein scaffold with HSV-1-induced AD | Aβ plaque formation, neuroinflammation, decreased functionality | [136] | |
| iPSCs-derived neuro-spheroids | Aβ aggregation; platform for testing of AD drugs | [137] | |
| 3D human neural progenitor cells | Show the importance of reducing the Aβ42/40 ratio for amelioration of AD; accurate tau pathology | [138] | |
| Acoustofluidic platform for assembly of neurospheroids and Aβ plaques | High throughput screening platform to test drugs against Aβ plaques | [139] | |
| 3D triculture of neurons, astrocytes, and microglial cells | Aβ aggregation, accumulation of p-tau, cytokine secretion | [140] | |
| In vivo models | APP overexpressing mice | Aβ plaque formation, learning, and cognitive deficits after 6 months | [141] |
| Aβ-GFP transgenic mice | Aβ is only able to form oligomers, thereby representing AD. Mice showed loss of memory, spine alterations, and increased p-tau levels. | [142] | |
| hTauP301L transgenic mice | Increased levels of phosphorylated tau, increased tau aggregation, neuronal loss | [143] | |
| T40PL-GFP transgenic mice, with the P301L 2N4R tau mutation | Increased levels of tau aggregation and tau pathology after 3 months | [144] | |
| ICV injection of Aβ oligomers | Memory loss in an ERK1/2-mediated fashion | [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. https://doi.org/10.3390/ijms232213954
Passeri E, Elkhoury K, Morsink M, Broersen K, Linder M, Tamayol A, Malaplate C, Yen FT, Arab-Tehrany E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. International Journal of Molecular Sciences. 2022; 23(22):13954. https://doi.org/10.3390/ijms232213954
Chicago/Turabian StylePasseri, Elodie, Kamil Elkhoury, Margaretha Morsink, Kerensa Broersen, Michel Linder, Ali Tamayol, Catherine Malaplate, Frances T. Yen, and Elmira Arab-Tehrany. 2022. "Alzheimer’s Disease: Treatment Strategies and Their Limitations" International Journal of Molecular Sciences 23, no. 22: 13954. https://doi.org/10.3390/ijms232213954
APA StylePasseri, E., Elkhoury, K., Morsink, M., Broersen, K., Linder, M., Tamayol, A., Malaplate, C., Yen, F. T., & Arab-Tehrany, E. (2022). Alzheimer’s Disease: Treatment Strategies and Their Limitations. International Journal of Molecular Sciences, 23(22), 13954. https://doi.org/10.3390/ijms232213954