
Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization

 , , , and
, , , and
Abstract
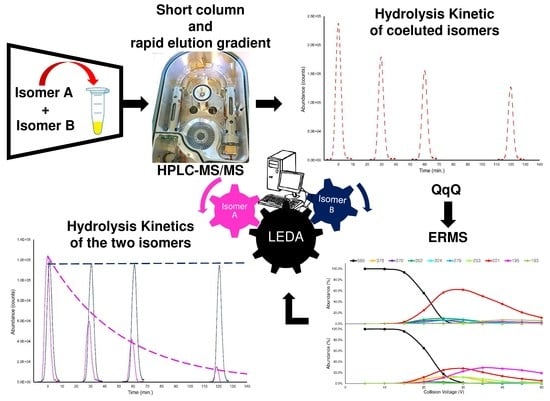
1. Introduction
2. Results
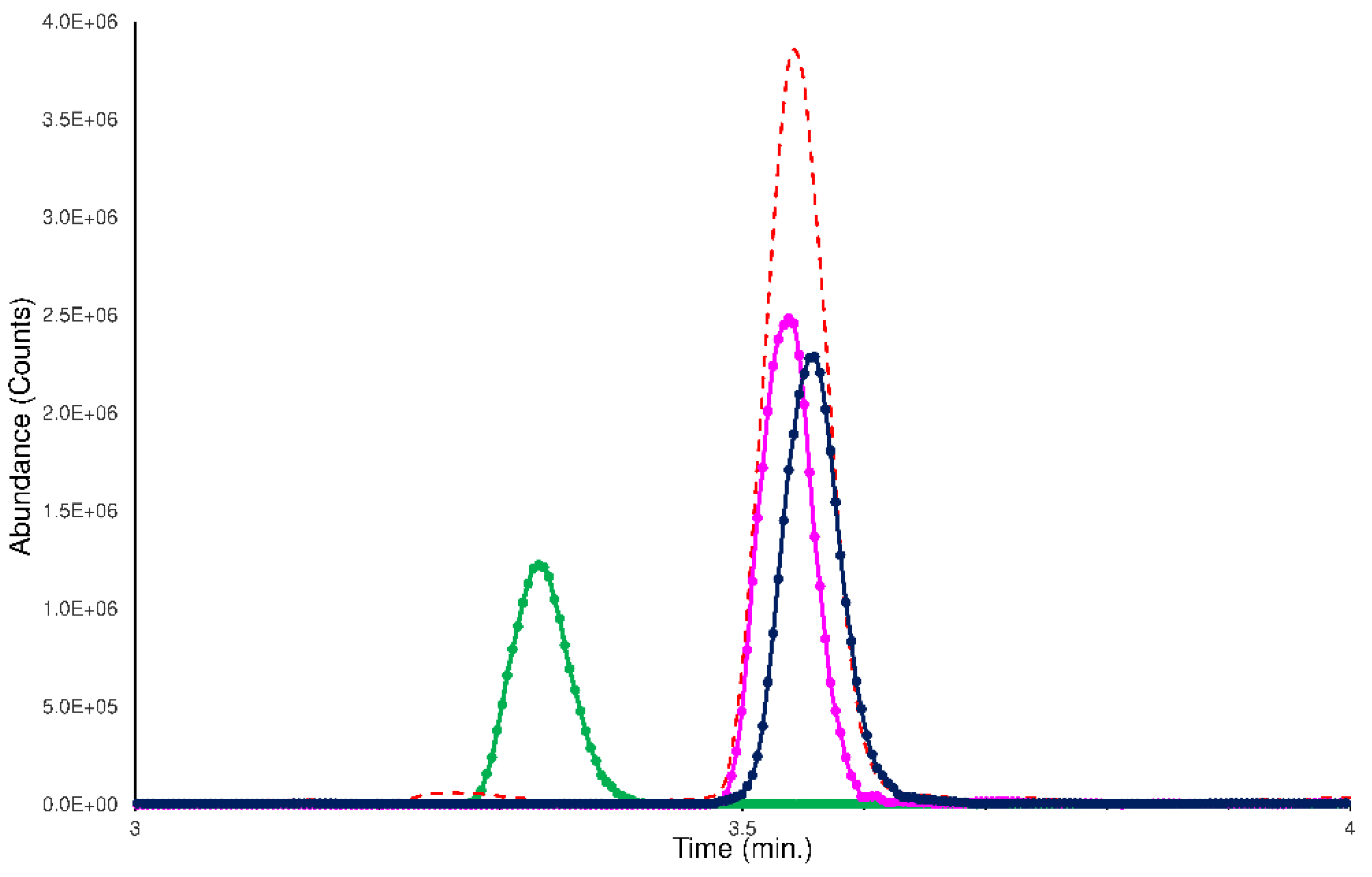
- the checking of the achieved chromatographic separation;
- the exploration of the MS/MS features in the isomers distinction;
- the evaluation of a mathematical device (LEDA) that allows the conversion of the common MS/MS signals in specific isomer abundances, related to their relative concentration in the sample;
- the assessment of the LEDA quantitative performances;
- the application of the optimized conditions of the LEDA approach in plasma stability experiments.
2.1. Chromatographic Separation
2.2. Collision-Induced Dissociation Sudy
2.3. LEDA Algorithm
- (Pi/Ri)m is the abundance ratio between the product ion (Pi) vs reference ion (Ri) measured (m) in the sample.
- (Pi/Ri)x is the characteristic abundance ratio between the Pi vs Ri of pure isomer.
- [%]x is the concentration (%) of the isomer in the sample.
2.4. LEDA Reliability
2.5. Evaluation of the Quantitative Performance of the MS/MS Analysis
2.6. Chemical Stability Test
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instrumental
4.3. Standard and CalibrationSsolutions
4.4. HPLC-MS/MS Methods
4.5. Sample Preparation
4.6. Validation of HPLC-MS/MS Methods
4.7. LEDA Algorithm
4.8. Chemical Stability Test
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lombardino, J.; Lowe, J. The role of the medicinal chemist in drug discovery—Then and now. Nat. Rev. Drug Discov. 2004, 3, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.B.; Macdonald, S.J.F.; Procopiou, P.A. Medicinal chemistry in drug discovery in big pharma: Past, present and future. Drug Discov. 2018, 23, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Korfmacher, W.A. Mass Spectrometry for Drug Discovery and Drug Development; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Wen, B.; Zhu, M. Applications of mass spectrometry in drug metabolism: 50 years of progress. Drug Metab. Rev. 2015, 47, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.; Shaffer, C.L.; Nedderman, A. Analytical strategies for identifying drug metabolites. Mass Spectrom. Rev. 2007, 26, 340–369. [Google Scholar] [CrossRef] [PubMed]
- Benassi, M.; Eberlin, M.N. Absolute Assignment of Constitutional Isomers via Structurally Diagnostic Fragment Ions: The Challenging Case of α- and β-Acyl Naphthalenes. J. Am. Soc. Mass Spectrom. 2010, 21, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Rathod, R.H.; Chaudhari, S.R.; Patil, A.S.; Shirkhedkar, A.A. Ultra-high performance liquid chromatography-MS/MS (UHPLC-MS/MS) in practice: Analysis of drugs and pharmaceutical formulations. Future J. Pharm Sci. 2019, 5, 6. [Google Scholar] [CrossRef]
- Ferrer, I.; Ruiz-Sala, P.; Vicente, Y.; Merinero, B.; Pérez-Cerdá, C.; Ugarte, M. Separation and identification of plasma short-chain acylcarnitine isomers by HPLC/MS/MS for the differential diagnosis of fatty acid oxidation defects and organic acidemias. J. Chromatogr. B 2007, 860, 121–126. [Google Scholar] [CrossRef]
- Benskin, J.P.; Ikonomou, M.G.; Woudneh, M.B.; Cosgrove, J.R. Rapid characterization of perfluoralkyl carboxylate, sulfonate, and sulfonamide isomers by high-performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2012, 1247, 165–170. [Google Scholar] [CrossRef]
- Karaźniewicz-Łada, M.; Danielak, D.; Teżyk, A.; Żaba, C.; Tuffal, G.; Główka, F. HPLC–MS/MS method for the simultaneous determination of clopidogrel, its carboxylic acid metabolite and derivatized isomers of thiol metabolite in clinical samples. J. Chromatogr. B 2012, 911, 105–112. [Google Scholar] [CrossRef]
- Crotti, S.; Menicatti, M.; Pallecchi, M.; Bartolucci, G. Tandem mass spectrometry approaches for recognition of isomeric compounds mixtures. Mass Spectrom. Rev. 2021, e21757. [Google Scholar] [CrossRef]
- Kurimoto, A.; Daikoku, S.; Mutsuga, S.; Kanie, O. Analysis of energy-resolved mass spectra at MSn in a Pursuit to characterize structural isomers of oligosaccharides. Anal. Chem. 2006, 78, 3461–3466. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, T.M.; Hall, L.H.; Hill, D.W.; Grant, D.F. CE50: Quantifying collision induced dissociation energy for small molecule characterization and identification. J. Am. Soc. Mass Spectrom. 2009, 20, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Memboeuf, A.; Jullien, L.; Lartia, R.; Brasme, B.; Gimbert, Y. Tandem mass spectrometric analysis of a mixture of isobars using the survival yield technique. J. Am. Soc. Mass Spectrom. 2011, 22, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Dit Fouque, J.D.; Maroto, A.; Memboeuf, A. Purification and Quantification of an Isomeric Compound in a Mixture by Collisional Excitation in Multistage Mass Spectrometry Experiments. Anal. Chem. 2016, 88, 10821–10825. [Google Scholar] [CrossRef] [PubMed]
- Dit Fouque, D.J.; Maroto, A.; Memboeuf, A. Internal Standard Quantification Using Tandem Mass Spectrometry of a Tryptic Peptide in the Presence of an Isobaric Interference. Anal. Chem. 2018, 90, 14126–14130. [Google Scholar] [CrossRef] [PubMed]
- Dit Fouque, J.D.; Lartia, R.; Maroto, A.; Memboeuf, A. Quantification of intramolecular click chemistry modified synthetic peptide isomers in mixtures using tandem mass spectrometry and the survival yield technique. Anal. Bioanal. Chem. 2018, 410, 5765–5777. [Google Scholar] [CrossRef]
- Maroto, A.; Dit Fouque, D.J.; Memboeuf, A. Ion trap MS using high trapping gas pressure enables unequivocal structural analysis of three isobaric compounds in a mixture by using energy-resolved mass spectrometry and the survival yield technique. J. Mass Spectrom. 2020, 55, e4478. [Google Scholar] [CrossRef]
- Dit Fouque, J.D.; Maroto, A.; Memboeuf, A. Structural analysis of a compound despite the presence of an isobaric interference by using in-source Collision Induced Dissociation and tandem mass spectrometry. J. Mass Spectrom. 2021, 56, e4698. [Google Scholar] [CrossRef]
- Campbell, M.T.; Chen, D.; Glish, G.L. Identifying the D-Pentoses Using Water Adduction to Lithium Cationized Molecule. J. Am. Soc. Mass Spectrom. 2017, 28, 1420–1424. [Google Scholar] [CrossRef]
- Campbell, M.T.; Chen, D.; Wallbillich, N.J.; Glish, G.L. Distinguishing Biologically Relevant Hexoses by Water Adduction to the Lithium-Cationized Molecule. Anal. Chem. 2017, 89, 10504–10510. [Google Scholar] [CrossRef]
- Campbell, M.T.; Chen, D.; Glish, G.L. Distinguishing Linkage Position and Anomeric Configuration of Glucose-Glucose Disaccharides by Water Adduction to Lithiated Molecules. Anal. Chem. 2018, 90, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- Menicatti, M.; Guandalini, L.; Dei, S.; Floriddia, E.; Teodori, E.; Traldi, P.; Bartolucci, G. The power of energy resolved tandem mass spectrometry experiments for resolution of isomers: The case of drug plasma stability investigation of multidrug resistance inhibitors. Rapid Commun. Mass Spectrom. 2016, 30, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Menicatti, M.; Guandalini, L.; Dei, S.; Floriddia, E.; Teodori, E.; Traldi, P.; Bartolucci, G. Energy resolved tandem mass spectrometry experiments for resolution of isobaric compounds: A case of cis/trans isomerism. Eur. J. Mass Spectrom. 2016, 22, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Menicatti, M.; Pallecchi, M.; Bua, S.; Vullo, D.; Di Cesare Mannelli, L.; Ghelardini, C.; Carta, F.; Supuran, C.T.; Bartolucci, G. Resolution of co-eluting isomers of anti-inflammatory drugs conjugated to carbonic anhydrase inhibitors from plasma in liquid chromatography by energy-resolved tandem mass spectrometry. J. Enzym. Inhib. Med. Chem. 2018, 33, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Menicatti, M.; Pallecchi, M.; Ricciutelli, M.; Galarini, R.; Moretti, S.; Sagratini, G.; Vittori, S.; Lucarini, S.; Caprioli, G.; Bartolucci, G. Determination of coeluted isomers in wine samples by application of MS/MS deconvolution analysis. J. Mass Spectrom. 2020, 55, e4607. [Google Scholar] [CrossRef]
- Pallecchi, M.; Menicatti, M.; Braconi, L.; Supuran, C.T.; Dei, S.; Teodori, E.; Bartolucci, G. Application of LEDA algorithm for the recognition of P-glycoprotein and Carbonic Anhydrase hybrid inhibitors and evaluation of their plasma stability by HPLC-MS/MS analysis. J. Pharm. Biomed. 2022, 219, 114887. [Google Scholar] [CrossRef]
- Dei, S.; Braconi, L.; Trezza, A.; Menicatti, M.; Contino, M.; Coronnello, M.; Chiaramonte, N.; Manetti, D.; Perrone, M.G.; Romanelli, M.N.; et al. Modulation of the spacer in N,N-bis(alkanol)amine aryl ester heterodimers led to the discovery of a series of highly potent P-glycoprotein-based multidrug resistance (MDR) modulators. Eur. J. Med. Chem. 2019, 172, 71–94. [Google Scholar] [CrossRef]
- Teodori, E.; Braconi, L.; Bua, S.; Lapucci, A.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Dei, S.; Supuran, C.T.; Coronnello, M. Dual P-Glycoprotein and CA XII Inhibitors: A New Strategy to Reverse the P-gp Mediated Multidrug Resistance (MDR) in Cancer Cells. Molecules 2020, 25, 1748. [Google Scholar] [CrossRef]
- Teodori, E.; Contino, M.; Riganti, C.; Bartolucci, G.; Braconi, L.; Manetti, D.; Romanelli, M.N.; Trezza, A.; Athanasios, A.; Spiga, O.; et al. Design, synthesis and biological evaluation of stereo- and regioisomers of amino aryl esters as multidrug resistance (MDR) reversers. Eur. J. Med. Chem. 2019, 182, 111655. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- ICH Q2B, Validation of Analytical Procedure: Methodology, International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. 1996. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 18 September 2022).
- Guandalini, L.; Soldani, G.; Rosi, L.; Calamai, L.; Bartolucci, G. A methodological approach to the selection of liquid reagents for chemical ionization ion trap-gas chromatography mass spectrometry: A case study of GBL and 1,4-BD. Int. J. Mass Spectrom. 2015, 388, 34–39. [Google Scholar] [CrossRef]








{kind=link}
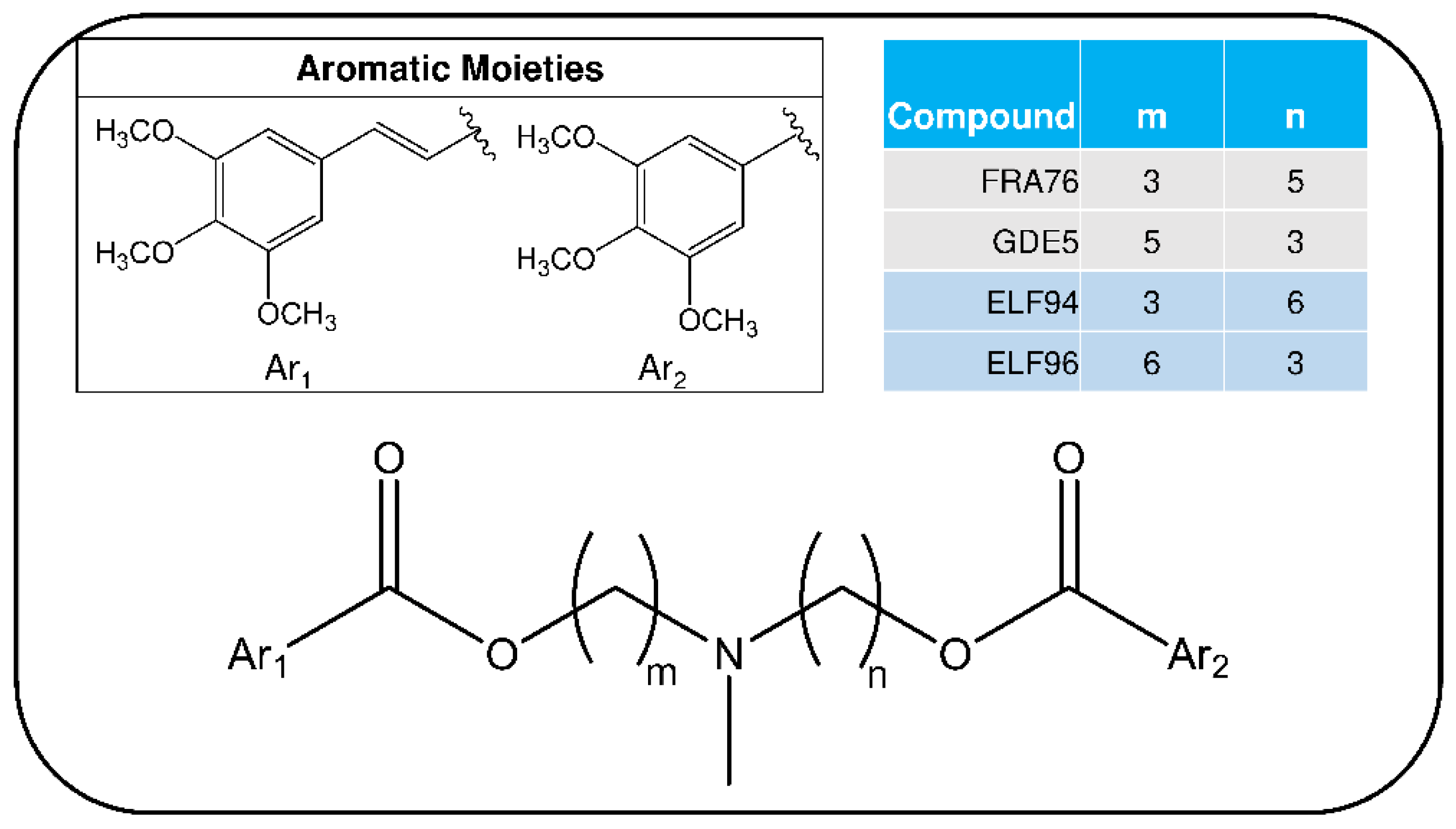{kind=link}
{kind=link}
{kind=link}
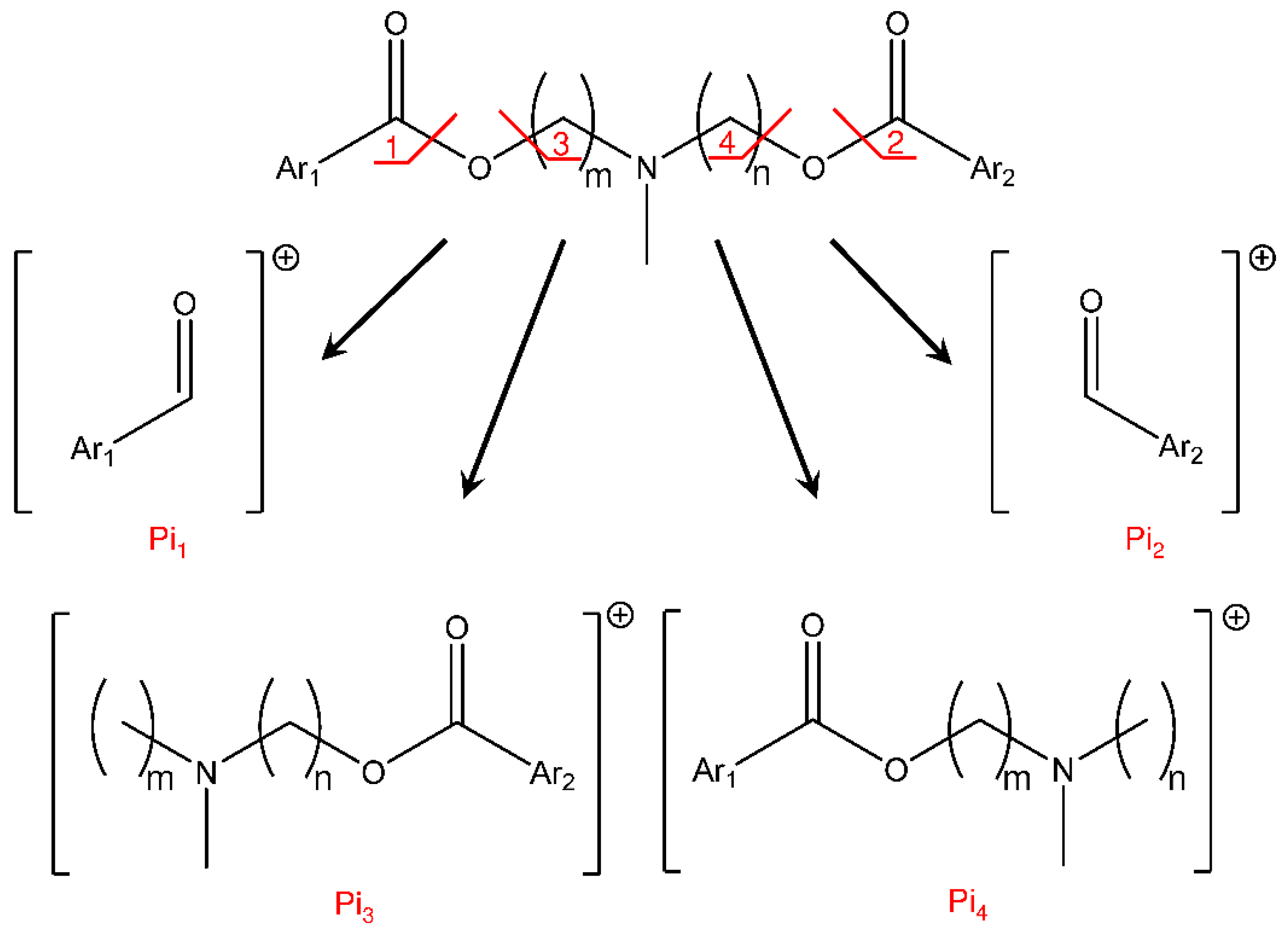{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slope | y-Intercept | R2 | SE-Lin | |
|---|---|---|---|---|
| FRA76 LEDA195–221 | 1.05 | −0.05 | 0.994 | 0.03 |
| FRA76 LEDAAll | 1.05 | −0.05 | 0.995 | 0.03 |
| GDE5 LEDA195–221 | 1.03 | 0.01 | 0.996 | 0.03 |
| GDE5 LEDAAll | 1.04 | 0.01 | 0.997 | 0.02 |
| ELF94 LEDA195–221 | 1.02 | −0.01 | 0.999 | 0.01 |
| ELF94 LEDAAll | 1.02 | −0.01 | 0.999 | 0.01 |
| ELF96 LEDA195–221 | 1.00 | −0.02 | 0.999 | 0.01 |
| ELF96 LEDAAll | 1.01 | −0.02 | 0.999 | 0.01 |
| Pure Isomer t1/2 ± 2SD (min) | Pure Isomer k ± 2SD (ln(μM)/min) | Mix t1/2 ± 2SD (min) | Mix k ± 2SD (ln(μM)/min) | |
|---|---|---|---|---|
| FRA76 | 43 ± 16 | −0.017 ± 0.001 | 32 ± 18 | −0.019 ± 0.003 |
| GDE5 | >120 | <0.006 | >120 | <0.006 |
| ELF94 | 49 ± 18 | −0.014 ± 0.001 | 34 ± 14 | −0.023 ± 0.005 |
| ELF96 | >120 | <0.006 | >120 | <0.006 |
| Compound | Precursor Ion (m/z) | MRM Signal | Product Ion (m/z) | CV (V) |
|---|---|---|---|---|
| IS | 455 | 165 | 30 | |
| FRA76 GDE5 | 590 | Ri | 590 (*) | 10 |
| Pi2 | 195 | 35 | ||
| Pi1 | 221 | 25 | ||
| Pi6 | 253 | 30 | ||
| Pi5 | 279 | 30 | ||
| Pi4 | 352 | 25 | ||
| Pi5 | 378 | 25 | ||
| ELF94 ELF96 | 604 | Ri | 604 (*) | 10 |
| Pi2 | 195 | 35 | ||
| Pi1 | 221 | 25 | ||
| Pi6 | 253 | 30 | ||
| Pi5 | 279 | 30 | ||
| Pi4 | 366 | 25 | ||
| Pi5 | 392 | 25 |
| Isomer Pair | Ratio Pi/Ri (m/z) | Isomer 1 Ratio Value ± SD | Isomer 2 Ratio Value ± SD |
|---|---|---|---|
| FRA76-GDE5 | 378/590 | 0.01 ± 0.01 | 0.26 ± 0.01 |
| 352/590 | 0.20 ± 0.01 | 0.04 ± 0.01 | |
| 279/590 | 0.11 ± 0.01 | 0.01 ± 0.01 | |
| 253/590 | 0.01 ± 0.01 | 0.21 ± 0.01 | |
| 221/590 | 1.30 ± 0.02 | 0.60 ± 0.01 | |
| 195/590 | 0.09 ± 0.01 | 0.64 ± 0.03 | |
| ELF94-ELF96 | 392/604 | 0.01 ± 0.01 | 0.32 ± 0.01 |
| 366/604 | 0.18 ± 0.01 | 0.01 ± 0.01 | |
| 279/604 | 0.08 ± 0.01 | 0.01 ± 0.01 | |
| 253/604 | 0.01 ± 0.01 | 0.18 ± 0.01 | |
| 221/604 | 1.10 ± 0.06 | 0.44 ± 0.03 | |
| 195/604 | 0.04 ± 0.01 | 0.70 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallecchi, M.; Braconi, L.; Menicatti, M.; Giachetti, S.; Dei, S.; Teodori, E.; Bartolucci, G. Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization. Int. J. Mol. Sci. 2022, 23, 13139. https://doi.org/10.3390/ijms232113139
Pallecchi M, Braconi L, Menicatti M, Giachetti S, Dei S, Teodori E, Bartolucci G. Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization. International Journal of Molecular Sciences. 2022; 23(21):13139. https://doi.org/10.3390/ijms232113139
Chicago/Turabian StylePallecchi, Marco, Laura Braconi, Marta Menicatti, Sara Giachetti, Silvia Dei, Elisabetta Teodori, and Gianluca Bartolucci. 2022. "Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization" International Journal of Molecular Sciences 23, no. 21: 13139. https://doi.org/10.3390/ijms232113139
APA StylePallecchi, M., Braconi, L., Menicatti, M., Giachetti, S., Dei, S., Teodori, E., & Bartolucci, G. (2022). Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization. International Journal of Molecular Sciences, 23(21), 13139. https://doi.org/10.3390/ijms232113139