
Cardiac Disease Alters Myocardial Tissue Levels of Epoxyeicosatrienoic Acids and Key Proteins Involved in Their Biosynthesis and Degradation

 ,
,  , ,
, , 
Abstract
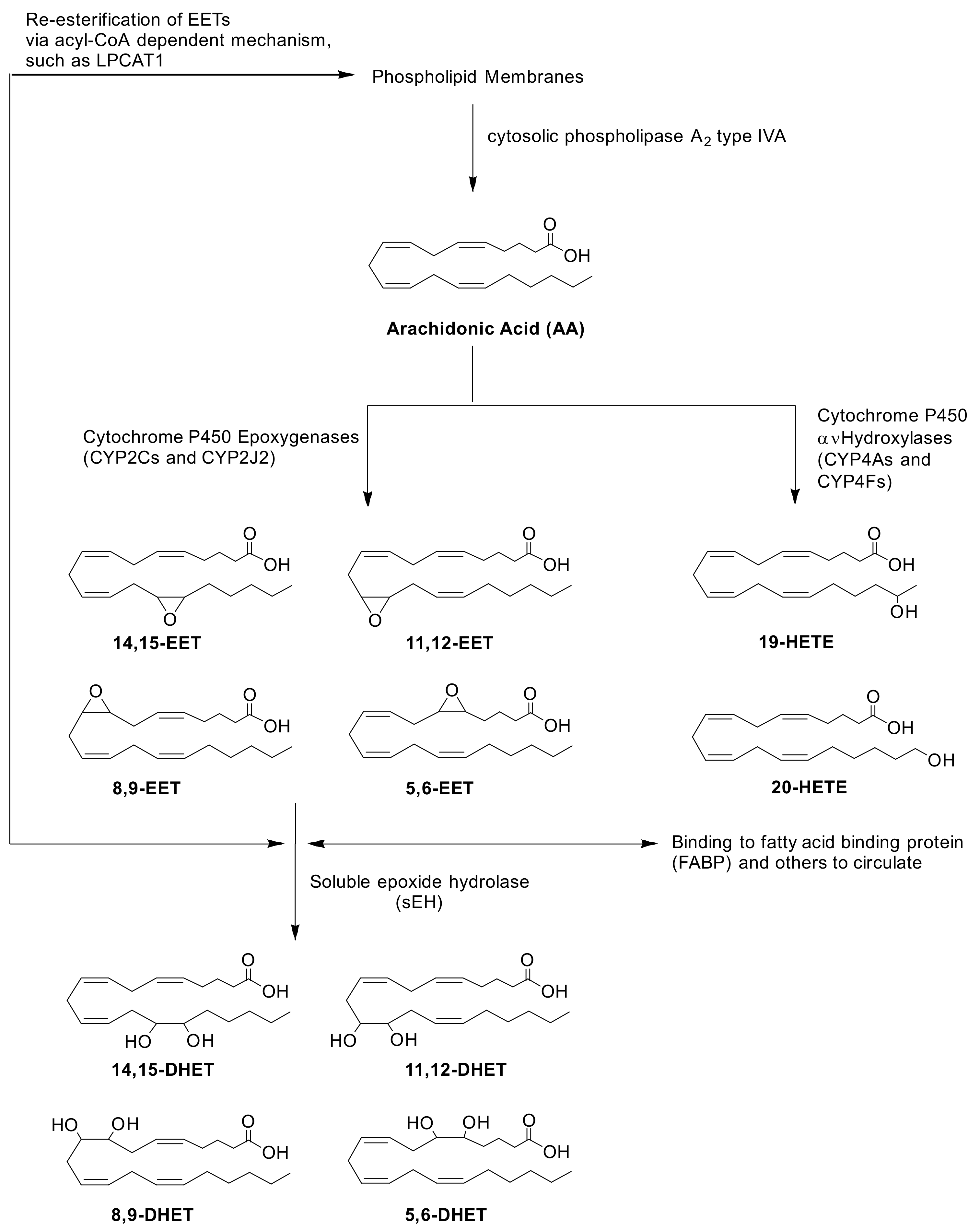
1. Introduction
2. Results
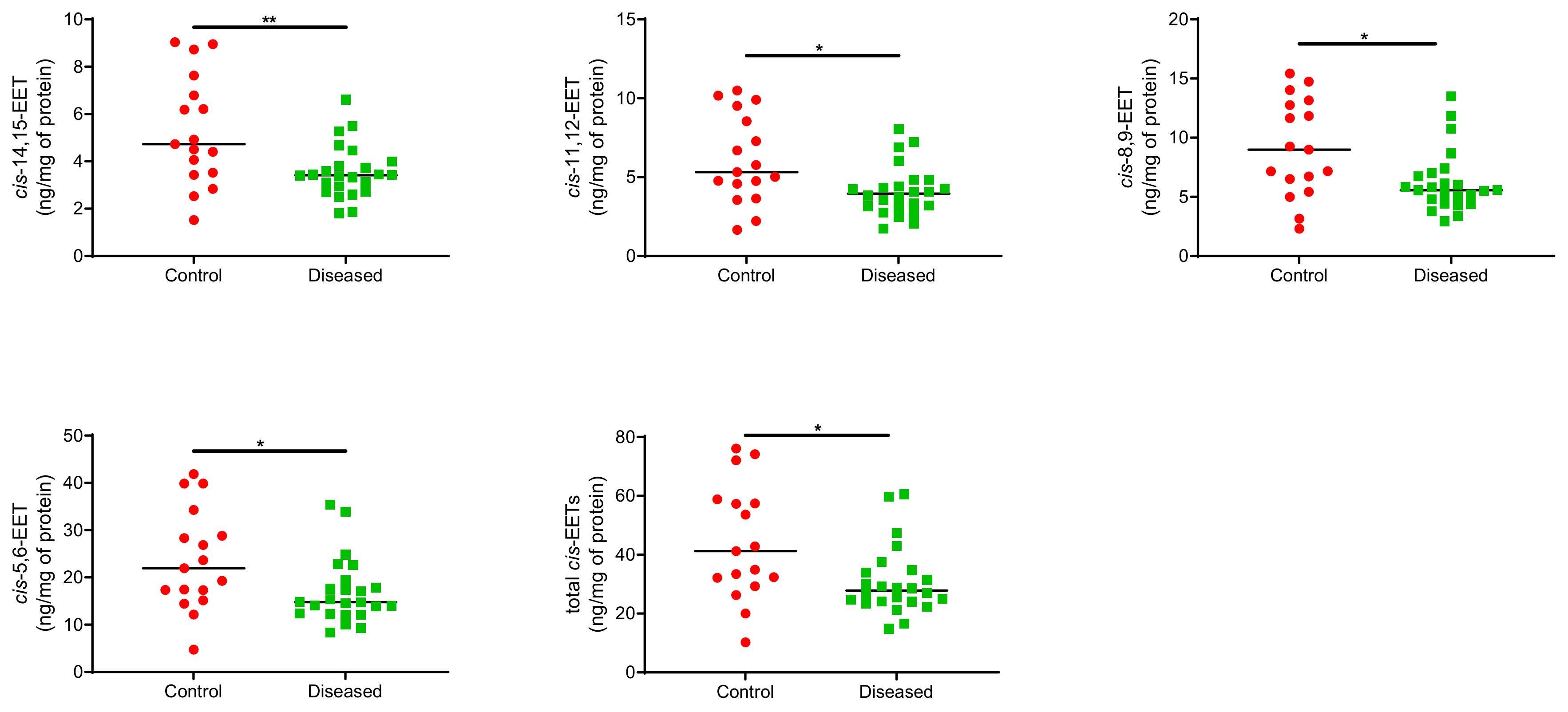
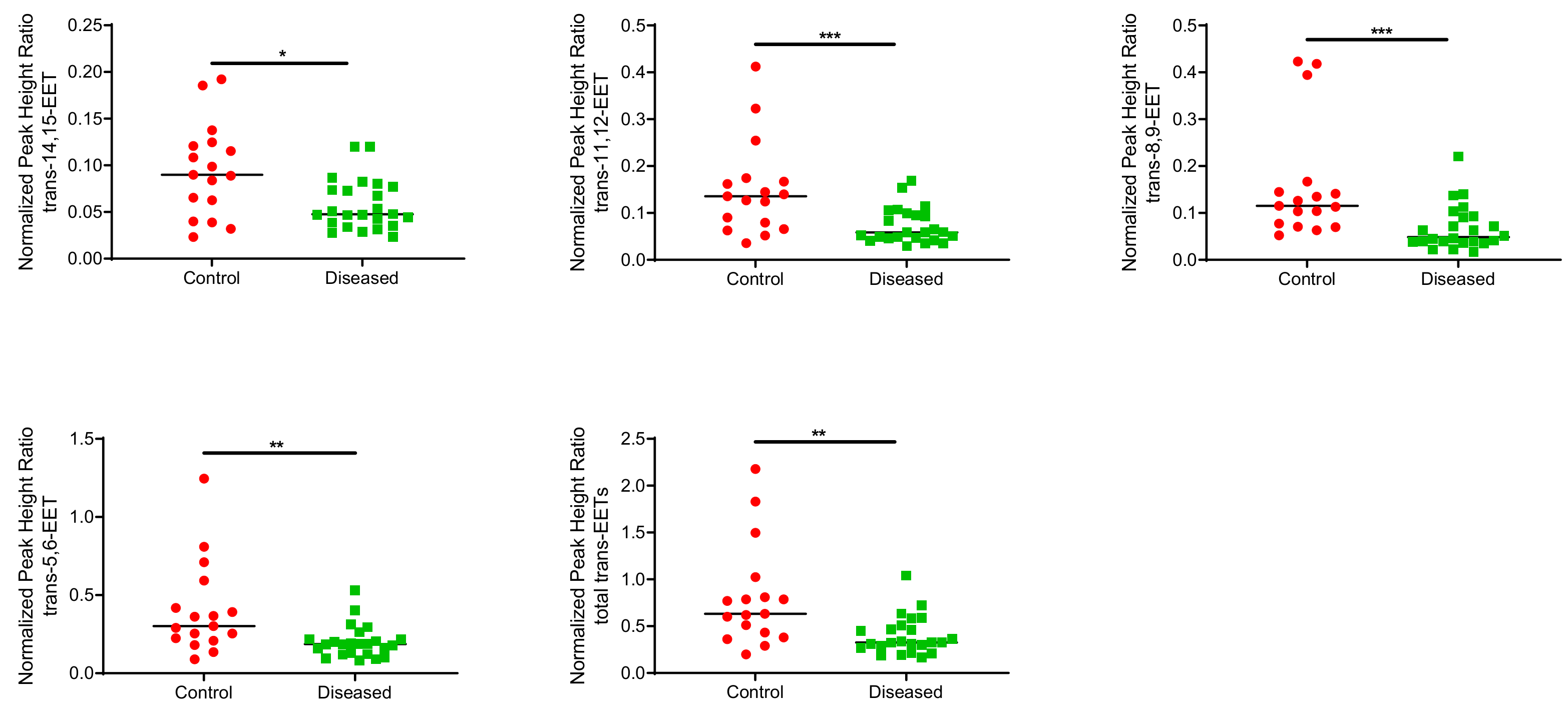
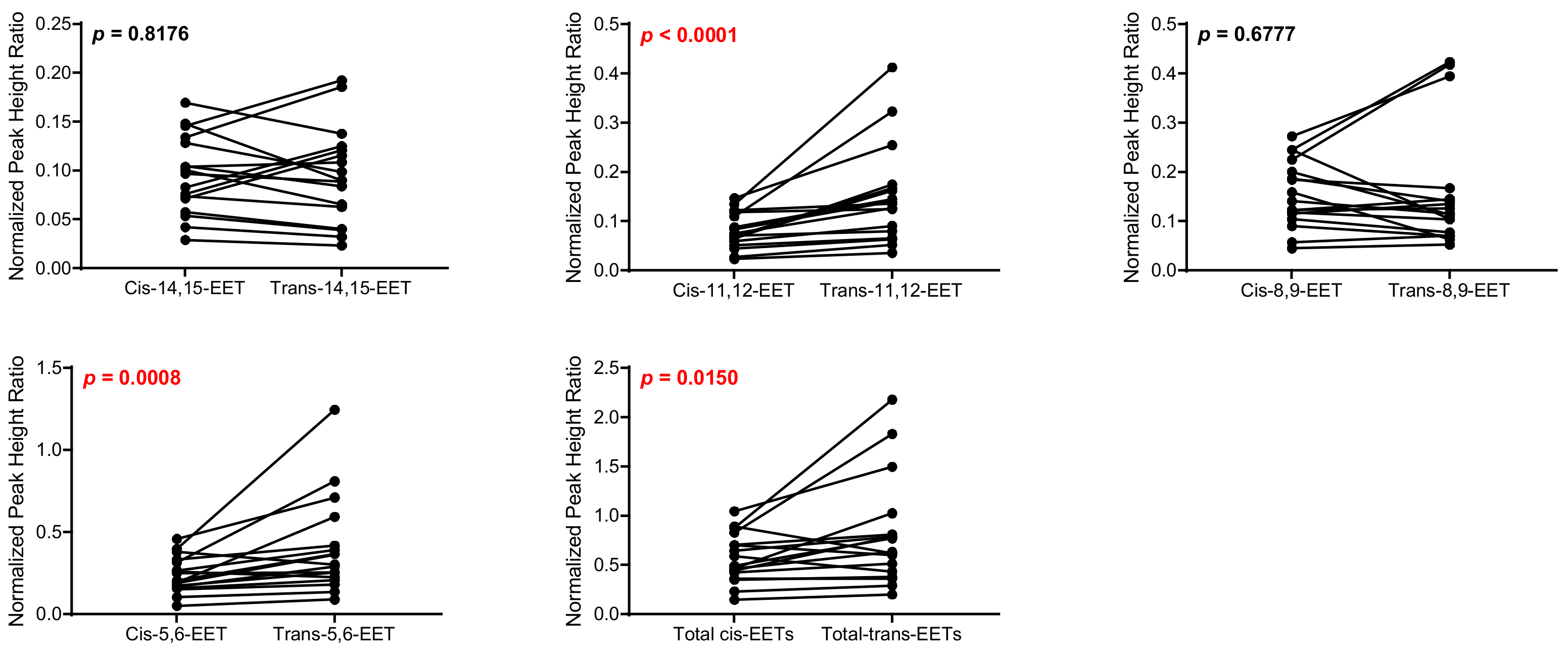
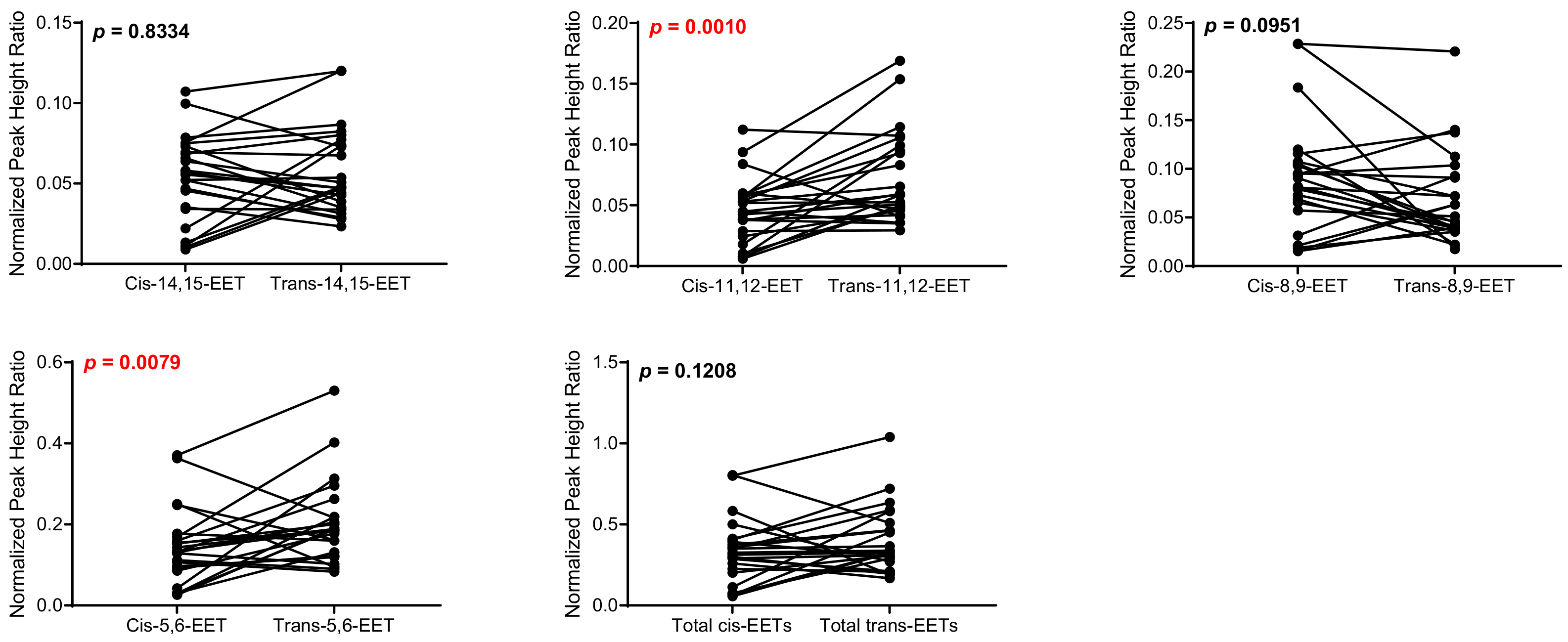
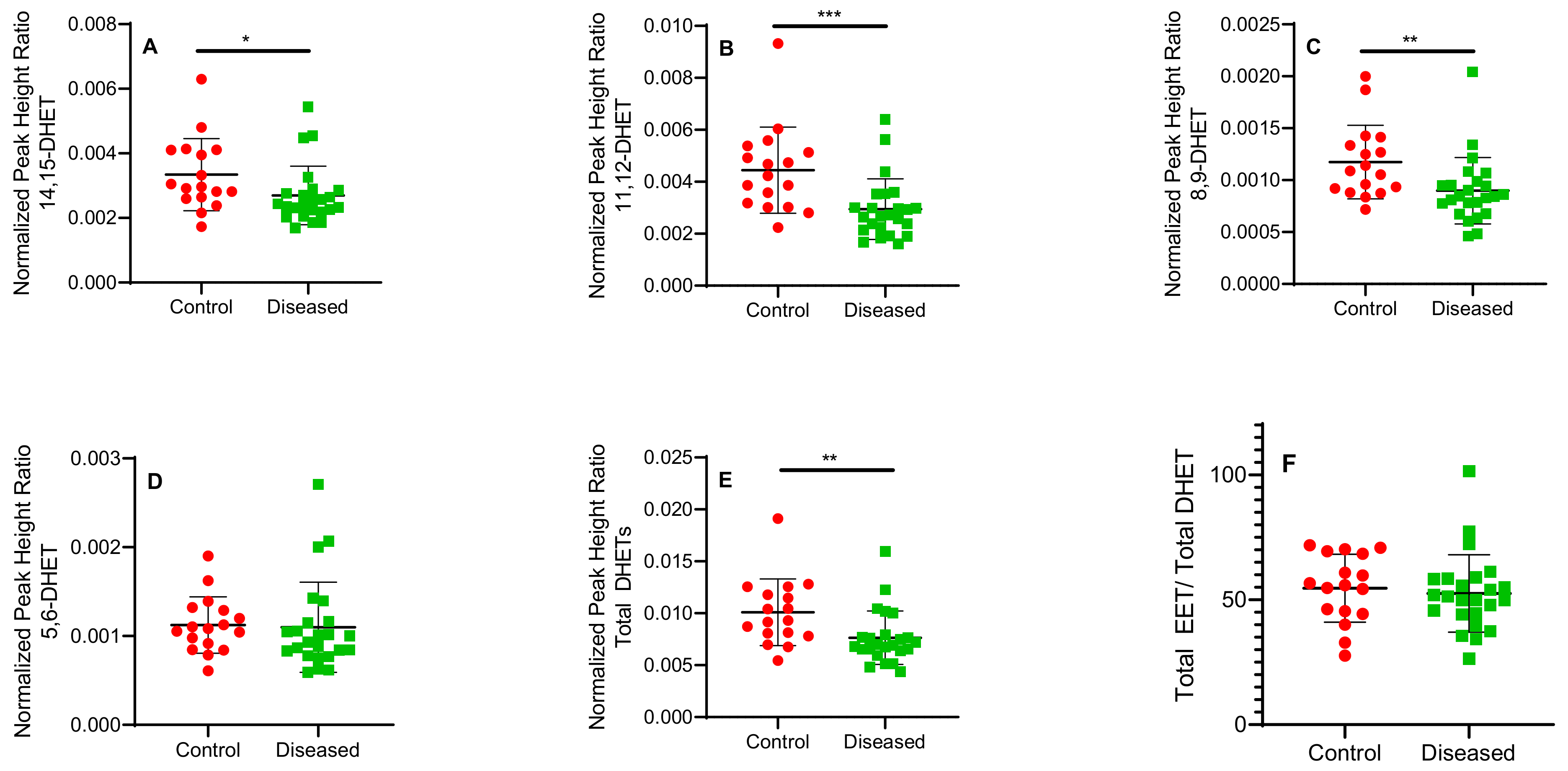
2.1. Total Free and Esterified EETs and DHETs from Human Cardiac Tissue
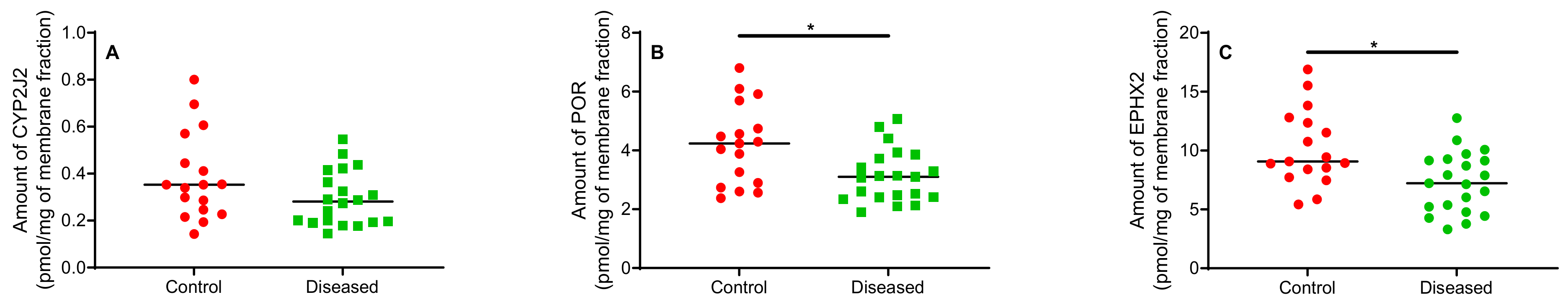
2.2. Protein Quantitation in Human Cardiac Tissue
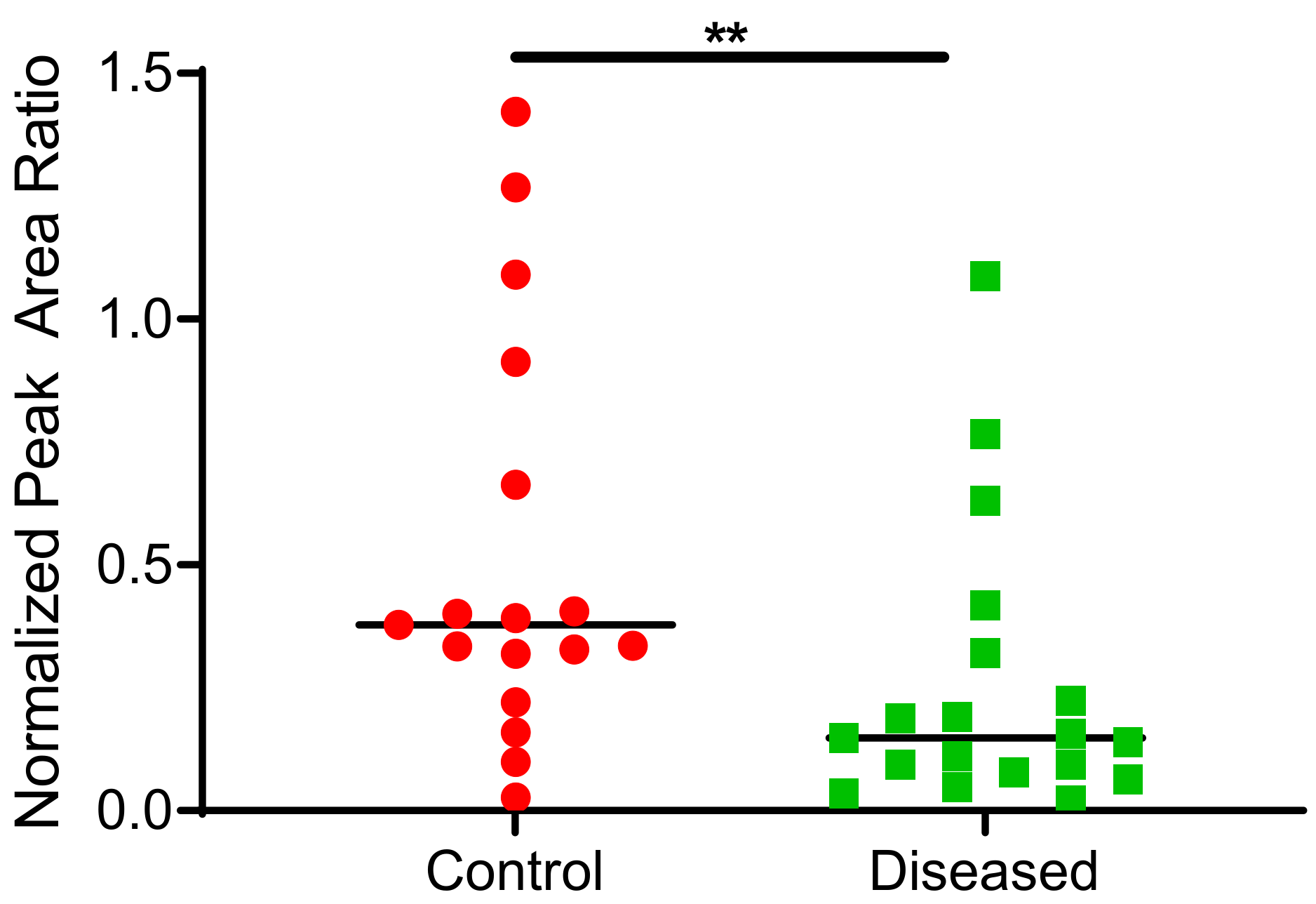
2.3. CYP2J2 Specific Activity in Human Cardiac Tissue Using Terfenadine as a Probe Substrate
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Study Samples
3.3. Quantitation of Total Free and Esterified EETs from Human Cardiac Tissue
3.4. Absolute and Relative Protein Quantitation
3.5. Protein Extraction and Trypsin Digestion
3.6. Mass Spectrometric Assay for Protein Quantitation
3.7. Metabolic Activity of CYP2J2 in Heart Tissue
3.8. LC-MS/MS Analysis of Terfenadine Oxidation in Heart Tissue
3.9. Data Analysis
3.10. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart disease and stroke statistics-2022 update: A report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.D.; Lin, L.L.; Kriz, R.W.; Ramesha, C.S.; Sultzman, L.A.; Lin, A.Y.; Milona, N.; Knopf, J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar] [CrossRef]
- Lin, L.L.; Wartmann, M.; Lin, A.Y.; Knopf, J.L.; Seth, A.; Davis, R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell 1993, 72, 269–278. [Google Scholar] [CrossRef]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar] [CrossRef]
- Karara, A.; Dishman, E.; Blair, I.; Falck, J.R.; Capdevila, J.H. Endogenous epoxyeicosatrienoic acids. Cytochrome P-450 controlled stereoselectivity of the hepatic arachidonic acid epoxygenase. J. Biol. Chem. 1989, 264, 19822–19827. [Google Scholar] [CrossRef]
- Goulitquer, S.; Dréano, Y.; Berthou, F.; Corcos, L.; Lucas, D. Determination of epoxyeicosatrienoic acids in human red blood cells and plasma by GC/MS in the NICI mode. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 876, 83–88. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, F.; Huse, L.M.; Morisseau, C.; Draper, A.J.; Newman, J.W.; Parker, C.; Graham, L.; Engler, M.M.; Hammock, B.D.; et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000, 87, 992–998. [Google Scholar] [CrossRef]
- Weintraub, N.L.; Fang, X.; Kaduce, T.L.; VanRollins, M.; Chatterjee, P.; Spector, A.A. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ. Res. 1997, 81, 258–267. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Johnson, C.O.; Hesselson, S.; Sotoodehnia, N.; Sotoodhenia, N.; McKnight, B.; Sitlani, C.M.; Rea, T.D.; King, I.B.; Kwok, P.Y.; et al. Common variation in fatty acid metabolic genes and risk of incident sudden cardiac arrest. Heart Rhythm 2014, 11, 471–477. [Google Scholar] [CrossRef]
- Moessinger, C.; Kuerschner, L.; Spandl, J.; Shevchenko, A.; Thiele, C. Human lysophosphatidylcholine acyltransferases 1 and 2 are located in lipid droplets where they catalyze the formation of phosphatidylcholine. J. Biol. Chem. 2011, 286, 21330–21339. [Google Scholar] [CrossRef]
- Akagi, S.; Kono, N.; Ariyama, H.; Shindou, H.; Shimizu, T.; Arai, H. Lysophosphatidylcholine acyltransferase 1 protects against cytotoxicity induced by polyunsaturated fatty acids. FASEB J. 2016, 30, 2027–2039. [Google Scholar] [CrossRef]
- Theken, K.N.; Schuck, R.N.; Edin, M.L.; Tran, B.; Ellis, K.; Bass, A.; Lih, F.B.; Tomer, K.B.; Poloyac, S.M.; Wu, M.C.; et al. Evaluation of cytochrome P450-derived eicosanoids in humans with stable atherosclerotic cardiovascular disease. Atherosclerosis 2012, 222, 530–536. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Edin, M.L.; Lee, J.A.; Wells, M.A.; Christensen, E.S.; Vendrov, K.C.; Lih, F.B.; Tomer, K.B.; Bai, X.; Taylor, J.M.; et al. Cytochrome P450-derived epoxyeicosatrienoic acids and coronary artery disease in humans: A targeted metabolomics study. J. Lipid. Res. 2016, 57, 109–119. [Google Scholar] [CrossRef]
- Minuz, P.; Jiang, H.; Fava, C.; Turolo, L.; Tacconelli, S.; Ricci, M.; Patrignani, P.; Morganti, A.; Lechi, A.; McGiff, J.C. Altered release of cytochrome p450 metabolites of arachidonic acid in renovascular disease. Hypertension 2008, 51, 1379–1385. [Google Scholar] [CrossRef]
- Monti, J.; Fischer, J.; Paskas, S.; Heinig, M.; Schulz, H.; Gösele, C.; Heuser, A.; Fischer, R.; Schmidt, C.; Schirdewan, A.; et al. Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat. Genet. 2008, 40, 529–537. [Google Scholar] [CrossRef]
- Deshane, J.; Wright, M.; Agarwal, A. Heme oxygenase-1 expression in disease states. Acta Biochim. Pol. 2005, 52, 273–284. [Google Scholar] [CrossRef]
- Evangelista, E.A.; Lemaitre, R.N.; Sotoodehnia, N.; Gharib, S.A.; Totah, R.A. CYP2J2 Expression in Adult Ventricular Myocytes Protects Against Reactive Oxygen Species Toxicity. Drug Metab. Dispos. 2018, 46, 380–386. [Google Scholar] [CrossRef]
- Yet, S.F.; Tian, R.; Layne, M.D.; Wang, Z.Y.; Maemura, K.; Solovyeva, M.; Ith, B.; Melo, L.G.; Zhang, L.; Ingwall, J.S.; et al. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ. Res. 2001, 89, 168–173. [Google Scholar] [CrossRef]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar] [CrossRef]
- Daniels, L.B.; Laughlin, G.A.; Sarno, M.J.; Bettencourt, R.; Wolfert, R.L.; Barrett-Connor, E. Lipoprotein-associated phospholipase A2 is an independent predictor of incident coronary heart disease in an apparently healthy older population: The Rancho Bernardo Study. J. Am. Coll. Cardiol. 2008, 51, 913–919. [Google Scholar] [CrossRef]
- Lee, C.A.; Neul, D.; Clouser-Roche, A.; Dalvie, D.; Wester, M.R.; Jiang, Y.; Jones, J.P.; Freiwald, S.; Zientek, M.; Totah, R.A. Identification of novel substrates for human cytochrome P450 2J2. Drug Metab. Dispos. 2010, 38, 347–356. [Google Scholar] [CrossRef]
- Basit, A.; Neradugomma, N.K.; Wolford, C.; Fan, P.W.; Murray, B.; Takahashi, R.H.; Khojasteh, S.C.; Smith, B.J.; Heyward, S.; Totah, R.A.; et al. Characterization of differential tissue abundance of major non-CYP enzymes in human. Mol. Pharm. 2020, 17, 4114–4124. [Google Scholar] [CrossRef]
- Aliwarga, T.; Raccor, B.S.; Lemaitre, R.N.; Sotoodehnia, N.; Gharib, S.A.; Xu, L.; Totah, R.A. Enzymatic and free radical formation of cis- and trans-epoxyeicosatrienoic acids in vitro and in vivo. Free Radic. Biol. Med. 2017, 112, 131–140. [Google Scholar] [CrossRef]
- Xu, M.; Bhatt, D.K.; Yeung, C.K.; Claw, K.G.; Chaudhry, A.S.; Gaedigk, A.; Pearce, R.E.; Broeckel, U.; Gaedigk, R.; Nickerson, D.; et al. Genetic and non-genetic factors associated with protein abundance of flavin-containing monooxygenase 3 in human liver. J. Pharmacol. Exp. Ther. 2017, 363, 265–274. [Google Scholar] [CrossRef]
- Evangelista, E.A.; Kaspera, R.; Mokadam, N.A.; Jones, J.P.; Totah, R.A. Activity, inhibition, and induction of cytochrome P450 2J2 in adult human primary cardiomyocytes. Drug Metab. Dispos. 2013, 41, 2087–2094. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.E.C.A.; Mathias, A.; et al. Abundance of phase 1 and 2 drug-metabolizing enzymes in alcoholic and hepatitis C cirrhotic livers: A quantitative targeted proteomics study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Heikkilä, P.; Laine, P.; Mattila, S.; Nieminen, M.S.; Parvinen, M.; Voipio-Pulkki, L.M. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur. J. Clin. Invest. 1999, 29, 380–386. [Google Scholar] [CrossRef]
- Marin-Garcia, J.; Goldenthal, M.J.; Moe, G.W. Mitochondrial pathology in cardiac failure. Cardiovasc. Res. 2001, 49, 17–26. [Google Scholar] [CrossRef]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Estabrook, R.W.; Franklin, M.R.; Cohen, B.; Shigamatzu, A.; Hildebrandt, A.G. Biochemical and genetic factors influencing drug metabolism. Influence of hepatic microsomal mixed function oxidation reactions on cellular metabolic control. Metabolism 1971, 20, 187–199. [Google Scholar] [CrossRef]
- Reed, J.R.; Cawley, G.F.; Backes, W.L. Inhibition of cytochrome P450 1A2-mediated metabolism and production of reactive oxygen species by heme oxygenase-1 in rat liver microsomes. Drug Metab. Lett. 2011, 5, 6–16. [Google Scholar] [CrossRef]
- Shen, A.L.; O’Leary, K.A.; Kasper, C.B. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem. 2002, 277, 6536–6541. [Google Scholar] [CrossRef]
- Fang, C.; Gu, J.; Xie, F.; Behr, M.; Yang, W.; Abel, E.D.; Ding, X. Deletion of the NADPH-cytochrome P450 reductase gene in cardiomyocytes does not protect mice against doxorubicin-mediated acute cardiac toxicity. Drug Metab. Dispos. 2008, 36, 1722–1728. [Google Scholar] [CrossRef]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef]
- Widstrom, R.L.; Norris, A.W.; Spector, A.A. Binding of cytochrome P450 monooxygenase and lipoxygenase pathway products by heart fatty acid-binding protein. Biochemistry 2001, 40, 1070–1076. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 14,15-EET | 11,12-EET | 8,9-EET | 5,6-EET | Total cis-EETs | |
|---|---|---|---|---|---|
| Control (n = 17) | 5.29 ± 2.3 | 6.11 ± 2.8 | 9.13 ± 4.1 | 23.7 ± 11 | 44.3 ± 20 |
| Diseased (n = 24) | 3.56 ± 1.1 | 4.12 ± 1.6 | 6.24 ± 2.6 | 16.9 ± 6.8 | 30.9 ± 12 |
| 14,15-EET | 11,12-EET | 8,9-EET | 5,6-EET | Total EETs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cis- | trans- | cis- | trans- | cis- | trans- | cis- | trans- | cis- | trans- | |
| Control (n = 17) | 0.0950 ± 0.040 | 0.0946 ± 0.05 | 0.0795 ± 0.046 | 0.0946 ± 0.05 | 0.155 ± 0.067 | 0.160 ± 0.12 | 0.238 ± 0.11 | 0.402 ± 0.29 | 0.567 ± 0.25 | 0.807 ± 0.55 |
| Diseased (n = 24) | 0.0533 ± 0.027 | 0.0575 ± 0.027 | 0.0453 ± 0.027 | 0.0725 ± 0.037 | 0.0906 ± 0.058 | 0.0683 ± 0.048 | 0.140 ± 0.093 | 0.201 ± 0.10 | 0.330 ± 0.20 | 0.399 ± 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliwarga, T.; Dinh, J.C.; Heyward, S.; Prasad, B.; Gharib, S.A.; Lemaitre, R.N.; Sotoodehnia, N.; Totah, R.A. Cardiac Disease Alters Myocardial Tissue Levels of Epoxyeicosatrienoic Acids and Key Proteins Involved in Their Biosynthesis and Degradation. Int. J. Mol. Sci. 2022, 23, 12433. https://doi.org/10.3390/ijms232012433
Aliwarga T, Dinh JC, Heyward S, Prasad B, Gharib SA, Lemaitre RN, Sotoodehnia N, Totah RA. Cardiac Disease Alters Myocardial Tissue Levels of Epoxyeicosatrienoic Acids and Key Proteins Involved in Their Biosynthesis and Degradation. International Journal of Molecular Sciences. 2022; 23(20):12433. https://doi.org/10.3390/ijms232012433
Chicago/Turabian StyleAliwarga, Theresa, Jean C. Dinh, Scott Heyward, Bhagwat Prasad, Sina A. Gharib, Rozenn N. Lemaitre, Nona Sotoodehnia, and Rheem A. Totah. 2022. "Cardiac Disease Alters Myocardial Tissue Levels of Epoxyeicosatrienoic Acids and Key Proteins Involved in Their Biosynthesis and Degradation" International Journal of Molecular Sciences 23, no. 20: 12433. https://doi.org/10.3390/ijms232012433
APA StyleAliwarga, T., Dinh, J. C., Heyward, S., Prasad, B., Gharib, S. A., Lemaitre, R. N., Sotoodehnia, N., & Totah, R. A. (2022). Cardiac Disease Alters Myocardial Tissue Levels of Epoxyeicosatrienoic Acids and Key Proteins Involved in Their Biosynthesis and Degradation. International Journal of Molecular Sciences, 23(20), 12433. https://doi.org/10.3390/ijms232012433