
Differential Analysis of Gly211Val and Gly286Val Mutations Affecting Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) in Congenital Pseudomyotonia Romagnola Cattle

 , ,
, ,  , , and
, , and 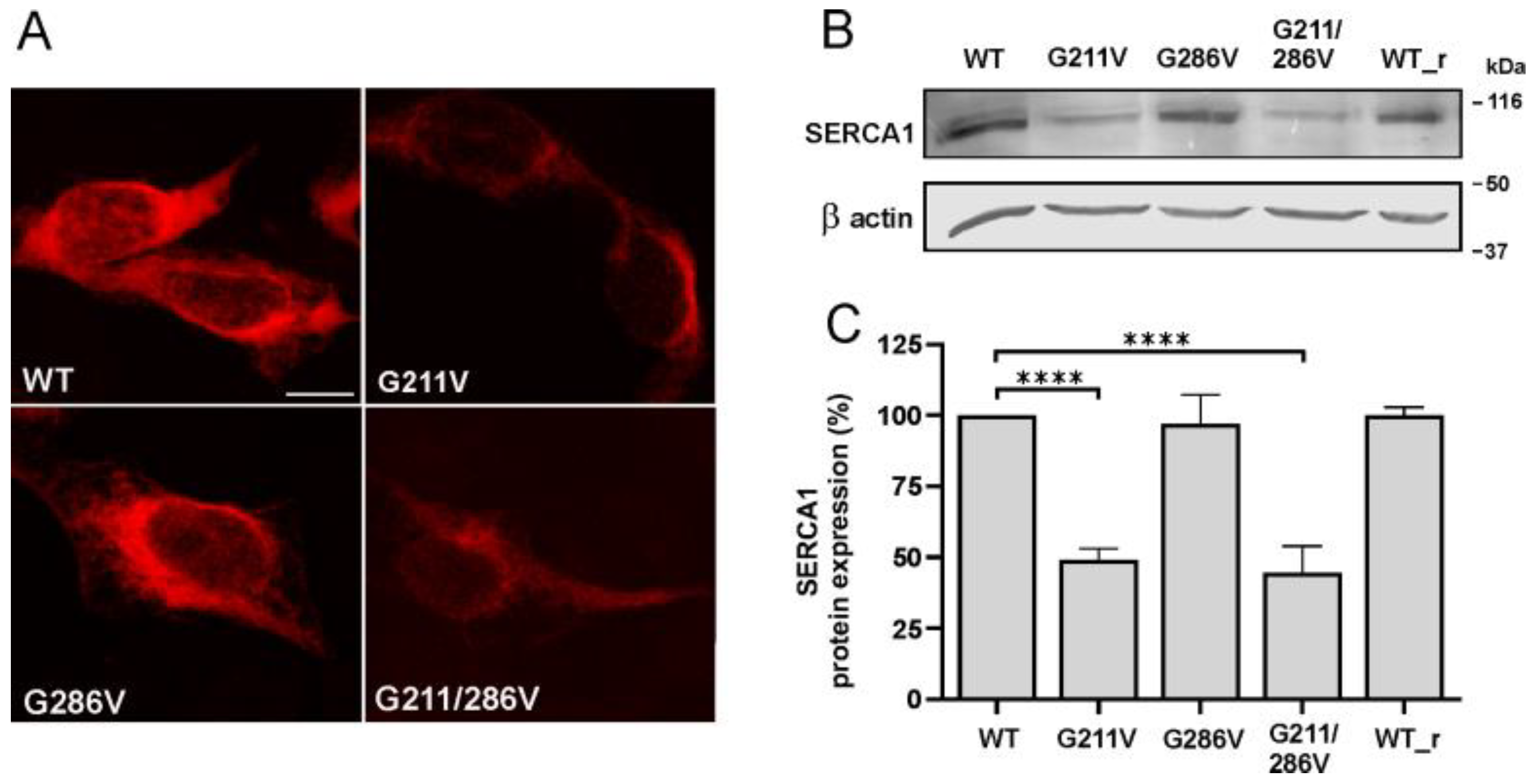{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of G211V and G286V Mutations on SERCA1 Protein Expression
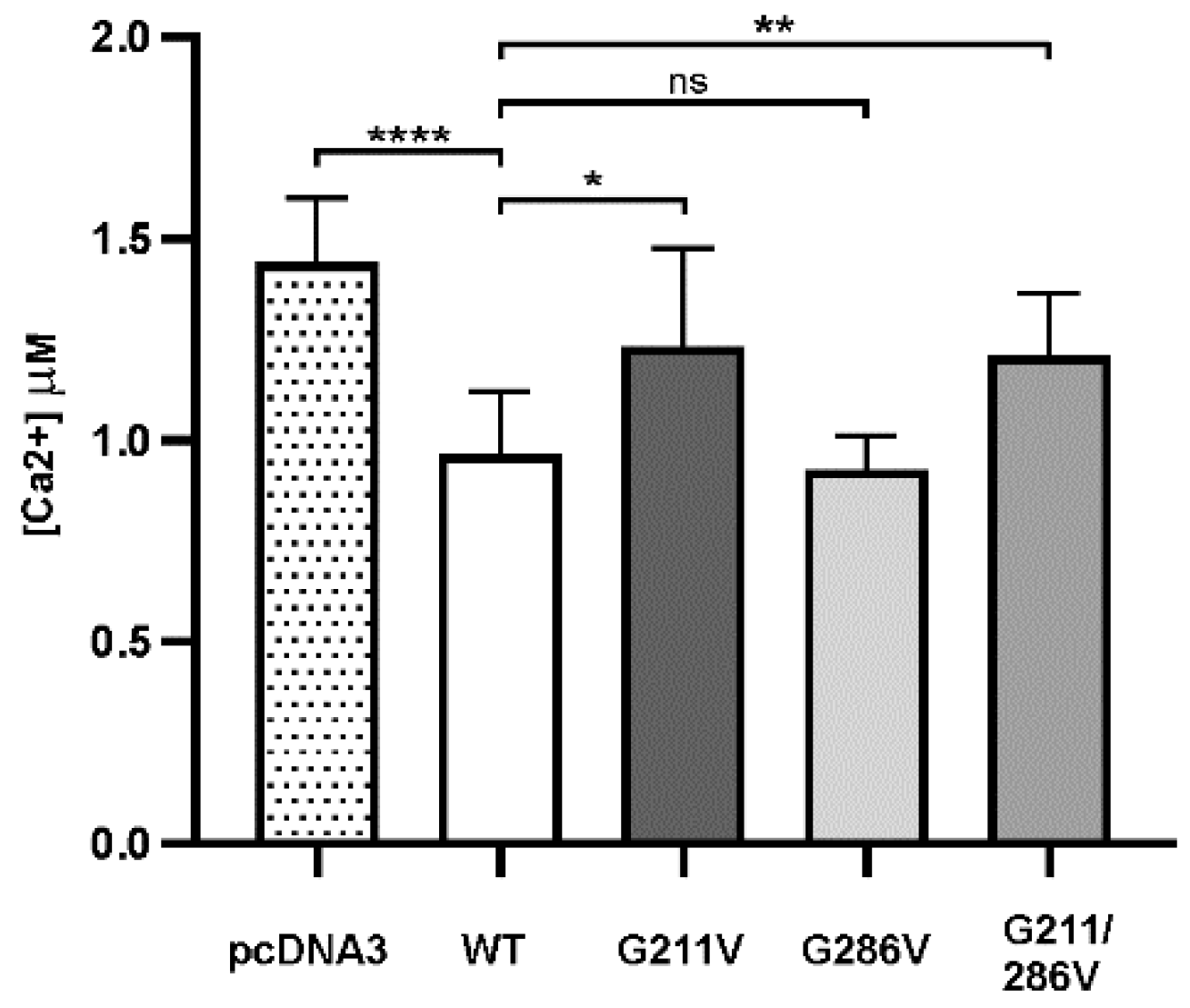
2.2. Functional Comparison of Cells Transfected with Different Vectors Expressing G211/286V, G211V and G286V Mutants
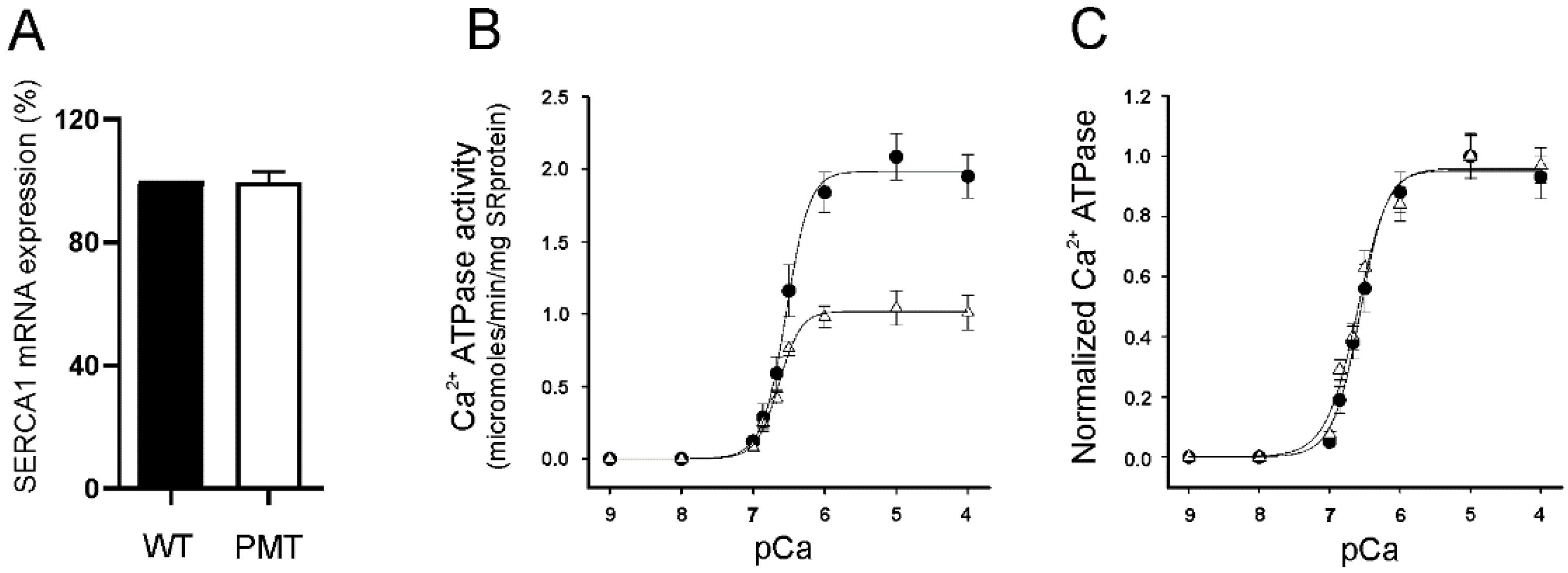
2.3. SERCA1 mRNA Expression Levels in Control and PMT-Affected Calf Muscles and Functional Comparison of Ca2+ ATPase Activities on Isolated Skeletal Muscle SR Membranes
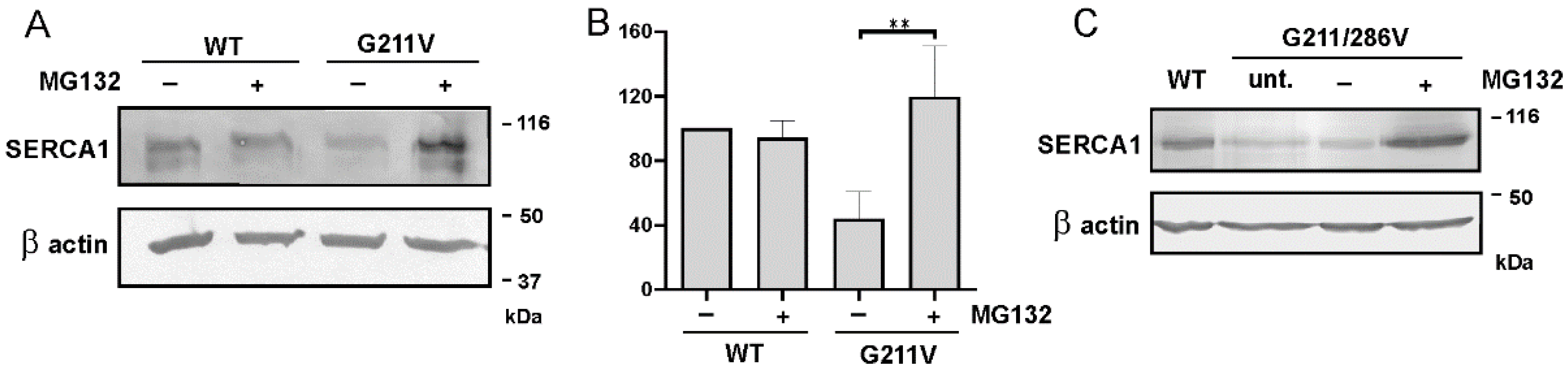
2.4. Quantitative and Qualitative Recovery of G211V SERCA1 Mutant after Proteasome Inhibition and Ubiquitination of G211V Mutant
3. Discussion
4. Materials and Methods
4.1. SERCA1 Construct and Site-Directed Mutagenesis
4.2. Cell Culture, Transfection and Treatment with Proteasome Inhibitor
4.3. Gel Electrophoresis and Immunoblotting
4.4. Immunofluorescence Analysis
4.5. Aequorin Ca2+ Measurements
4.6. Preparative Procedures and Biochemical Assay
4.7. Immunoprecipitation
4.8. RNA Preparation and Quantitative Real-Time PCR
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Testoni, S.; Boni, P.; Gentile, A. Congenital pseudomyotonia in Chianina cattle. Vet. Rec. 2008, 163, 252. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Coppieters, W.; Rollin, F.; Desmecht, D.; Agerholm, J.S.; Cambisano, N.; Carta, E.; Dardano, S.; Dive, M.; Fasquelle, C.; et al. Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat. Genet. 2008, 40, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Sacchetto, R.; Testoni, S.; Dorotea, T.; Mascarello, F.; Liguori, R.; Gentile, A.; Drögemüller, C. Pseudomyotonia in Romagnola cattle caused by novel ATP2A1 mutations. BMC Vet. Res. 2012, 8, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Dorotea, T.; Grünberg, W.; Murgiano, L.; Plattet, P.; Drögemüller, C.; Mascarello, F.; Sacchetto, R. Fast-twitch skeletal muscle fibers adaptation to SERCA1 fediciency in a Dutch Improved Red and White calf pseudomyotonia case. Neuromuscul. Disord. 2015, 25, 888–897. [Google Scholar] [CrossRef]
- Drögemüller, C.; Drögemüller, M.; Leeb, T.; Mascarello, F.; Testoni, S.; Rossi, M.; Gentile, A.; Damiani, E.; Sacchetto, R. Identification of a missense mutation in the bovine ATP2A1 gene in congenital pseudomyotonia of Chianina cattle: An animal model of human Brody disease. Genomics 2008, 92, 482–491. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, S.; Inui, M. Biochemistry and biophysics of excitation-contraction coupling. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 333–364. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, R.; Testoni, S.; Gentile, A.; Damiano, E.; Rossi, M.; Liguori, R.; Drögemüller, C.; Mascarello, F. A defective SERCA1 protein is responsible for congenital pseudomyotonia in Chianina cattle. Am. J. Pathol. 2009, 174, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, E.; Testoni, S.; Gentile, A.; Calì, T.; Ottolini, D.; Villa, A.; Brini, M.; Betto, R.; Mascarello, F.; Nissen, P.; et al. Inhibition of Ubiquitin Proteasome System Rescues the Defective Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) Protein Causing Chianina Cattle Pseudomyotonia. J. Biol. Chem. 2014, 289, 33073–33082. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Brandl, C.J.; Korczak, B.; Green, N.M. Amino-acid sequence of a Ca2+ + Mg2+-dependent ATPase from rabbit muscle sarcoplasmic reticulum, deduced from its complementary DNA sequence. Nature 1985, 316, 696–700. [Google Scholar] [CrossRef]
- Maruyama, K.; MacLennan, D.H. Mutation of aspartic acid-351, lysine-352, and lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Mascarello, F.; Sacchetto, R. Structural study of skeletal muscle fibres in healthy and pseudomyotonia affected cattle. Ann. Anat. 2016, 207, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Marsault, R.; Bastianutto, C.; Alvarez, J.; Pozzan, T.; Rizzuto, R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]). A critical evaluation. J. Biol. Chem. 1995, 270, 9896–9903. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Hu, Z.; Blackwell, D.J.; Miller, T.D.; Thomas, D.D.; Robia, S.L. 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS ONE 2012, 7, e40369. [Google Scholar] [CrossRef] [PubMed]
- Brody, I.A. Muscle contracture induced by exercise: A syndrome attributable to decreased relaxing factor. N. Engl. J. Med. 1996, 281, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Karpati, G.; Charuk, J.; Carpenter, S.; Jablecki, C.; Holland, P. Myopathy caused by a deficiency of Ca(2+)-adenosine triphosphatase in sarcoplasmic reticulum (Brody’s disease). Ann. Neurol. 1986, 20, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A.; Taschner, P.E.M.; Khanna, V.K.; Busch, H.F.M.; Karpati, G.; Jablecki, C.K.; Breuning, M.H.; MacLennan, D.H. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca(2+) ATPase, are associated with Brody disease. Nat. Genet. 1996, 14, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.P.; Verhoeven, J.I.; Rodenburg, R.J.; Kamsteeg, E.J.; Erasmus, C.E.; Vicart, S.; Behin, A.; Bassez, G.; Magot, A.; Pereon, Y.; et al. Clinical, morphological and genetic characterization of Brody disease: An international study of 40 patients. Brain 2020, 143, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Benders, A.A.G.M.; Veerkamp, J.H.; Oosterhof, A.; Jongen, P.J.H.; Bindels, R.J.M.; Smit, L.M.E.; Busch, H.F.M.; Wevers, R.A. Ca(2+) homeostasis in Brody’s disease: A study in skeletal muscle and cultured muscle cells and the effects of dantrolene and verapamil. J. Clin. Investig. 1994, 94, 741–748. [Google Scholar] [CrossRef]
- Sacchetto, R.; Bertipaglia, I.; Giannetti, S.; Cendron, L.; Mascarello, F.; Damiani, E.; Carafoli, E.; Zanotti, G. Crystal structure of sarcoplasmic reticulum Ca2+-ATPase (SERCA) from bovine muscle. J. Struct. Biol. 2012, 178, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, Y.; Daiho, T.; Yamasaki, K.; Takahashi, H.; Ishida-Yamamoto, A.; Danko, H.; Lizuka, H. Comprehensive analysis of expression and function of 51 sarco(endo)plasmic reticulum Ca2+-ATPase mutants associated with Darier disease. J. Biol. Chem. 2006, 281, 22882–22895. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Carter, S.A.; Healy, E.; Todd, C.; Rees, J.L.; Steijlen, P.M.; Carmichael, A.J.; Lewis, H.M.; Hohl, D.; Itin, P.; et al. ATP2A2 mutations in Darier’s disease: Variant cutaneous phenotypes are associated with missense mutations, but neuropsychiatric features are independent of mutation class. Hum. Mol. Genet. 1999, 8, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Molinski, S.; Eckford, P.D.; Pasy, S.; Ahmadi, S.; Chin, S.; Bear, C.E. Functional rescue of F508del-CFTR using small molecule correctors. Front. Pharmacol. 2012, 3, 160. [Google Scholar] [CrossRef]
- Carotti, M.; Marsolier, J.; Soardi, M.; Bianchini, E.; Gomiero, C.; Fecchio, C.; Henriques, S.F.; Betto, R.; Sacchetto, R.; Richard, I.; et al. Repairing folding-defective α-sarcoglycan mutants by CFTR correctors, a potential therapy for Limb Girdle Muscular Dystrophy 2D. Hum. Mol. Genet. 2018, 27, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Scano, M.; Benetollo, A.; Nogara, L.; Bondi, M.; Dalla Barba, F.; Soardi, M.; Furlan, S.; Akyurek, E.E.; Caccin, P.; Carotti, M.; et al. CFTR corrector C17 is effective in muscular dystrophy, in vivo proof of concept in LGMDR3. Hum. Mol. Genet. 2022, 31, 499–509. [Google Scholar] [CrossRef]
- Ward, C.L.; Satoshi, O.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef]
- Gastaldello, S.; D’Angelo, S.; Franzoso, S.; Fanin, M.; Angelini, C.; Betto, R.; Sandona, D. Inhibition of proteasome activity promotes the correct localization of disease-causing α-sarcoglycan mutants in HEK-293 cells constitutively expressing β-, γ-, and δ-sarcoglycan. Am. J. Pathol. 2008, 173, 170–181. [Google Scholar] [CrossRef]
- Pan, Y.; Zvaritch, E.; Tupling, A.R.; Rice, W.J.; de Leon, S.; Rudnicki, M.; McKerlie, C.; Banwell, B.L.; MacLennan, D.H. Targeted disruption of the ATP2A1 gene encoding the sarco(endo)plasmic reticulum Ca2+ ATPase isoform 1 (SERCA1) impairs diaphragm function and is lethal in neonatal mice. J. Biol. Chem. 2003, 278, 13367–13375. [Google Scholar] [CrossRef]
- Prasad, V.; Okunade, G.W.; Miller, M.L.; Shull, G.E. Phenotypes of SERCA and PMCA knockout mice. Biochem. Biophys. Res. Commun. 2004, 322, 1192–1203. [Google Scholar] [CrossRef]
- Shull, G.E.; Okunade, G.; Liu, L.; Kozel, P.; Periasamy, M.; Lorenz, J.N.; Prasad, V. Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Ann. N. Y. Acad. Sci. 2003, 986, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Bocchi, L.; Sacchetto, R.; Maria Cristina Florio, M.C.; Motta, B.M.; Corti, C.; Weichenberger, C.X.; Savi, M.; D’Elia, Y.; Rosato-Siri, M.D.; et al. HDAC inhibition improves the sarcoendoplasmic reticulum Ca2+-ATPase activity in cardiac myocytes. Int. J. Mol. Sci. 2018, 19, 419. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akyürek, E.E.; Busato, F.; Murgiano, L.; Bianchini, E.; Carotti, M.; Sandonà, D.; Drögemüller, C.; Gentile, A.; Sacchetto, R. Differential Analysis of Gly211Val and Gly286Val Mutations Affecting Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) in Congenital Pseudomyotonia Romagnola Cattle. Int. J. Mol. Sci. 2022, 23, 12364. https://doi.org/10.3390/ijms232012364
Akyürek EE, Busato F, Murgiano L, Bianchini E, Carotti M, Sandonà D, Drögemüller C, Gentile A, Sacchetto R. Differential Analysis of Gly211Val and Gly286Val Mutations Affecting Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) in Congenital Pseudomyotonia Romagnola Cattle. International Journal of Molecular Sciences. 2022; 23(20):12364. https://doi.org/10.3390/ijms232012364
Chicago/Turabian StyleAkyürek, Eylem Emek, Francesca Busato, Leonardo Murgiano, Elisa Bianchini, Marcello Carotti, Dorianna Sandonà, Cord Drögemüller, Arcangelo Gentile, and Roberta Sacchetto. 2022. "Differential Analysis of Gly211Val and Gly286Val Mutations Affecting Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) in Congenital Pseudomyotonia Romagnola Cattle" International Journal of Molecular Sciences 23, no. 20: 12364. https://doi.org/10.3390/ijms232012364
APA StyleAkyürek, E. E., Busato, F., Murgiano, L., Bianchini, E., Carotti, M., Sandonà, D., Drögemüller, C., Gentile, A., & Sacchetto, R. (2022). Differential Analysis of Gly211Val and Gly286Val Mutations Affecting Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) in Congenital Pseudomyotonia Romagnola Cattle. International Journal of Molecular Sciences, 23(20), 12364. https://doi.org/10.3390/ijms232012364