2.1.1. Derivatization
As it was envisioned, TCA did not possess the desired physicochemical properties to meet the requirements of the implemented technique. Therefore, chemical modification was a necessary part of the sample preparation process. Interestingly, a wide variety of derivatization methods have been developed to overcome the above-mentioned constraints. Among others, four types of derivatization reactions preceding a GC analysis are most commonly used, namely silylation, acylation, alkylation and esterification methods, to enable analysis of organic compounds with inadequate volatility or thermal stability; as well as to improve their chromatographic behavior or detectability [
20,
21,
22]. In the presented study, considerable attention was paid to the chemical reactions proceeding in media with high water content at room temperature in order to simplify the sample pretreatment procedure and improve batch to batch reproducibility; apart from increasing TCA hydrophobicity before the extraction step, as stated previously. Based on our previous experience with the development of the GC–MS method for urinary HTL determination [
23], the use of IBCF was evaluated. It was demonstrated that with the proper use of the above-mentioned derivatization reagent, a successful analysis of a wide range of organic compounds from environmental samples can be achieved [
19,
24].
Figure 1.
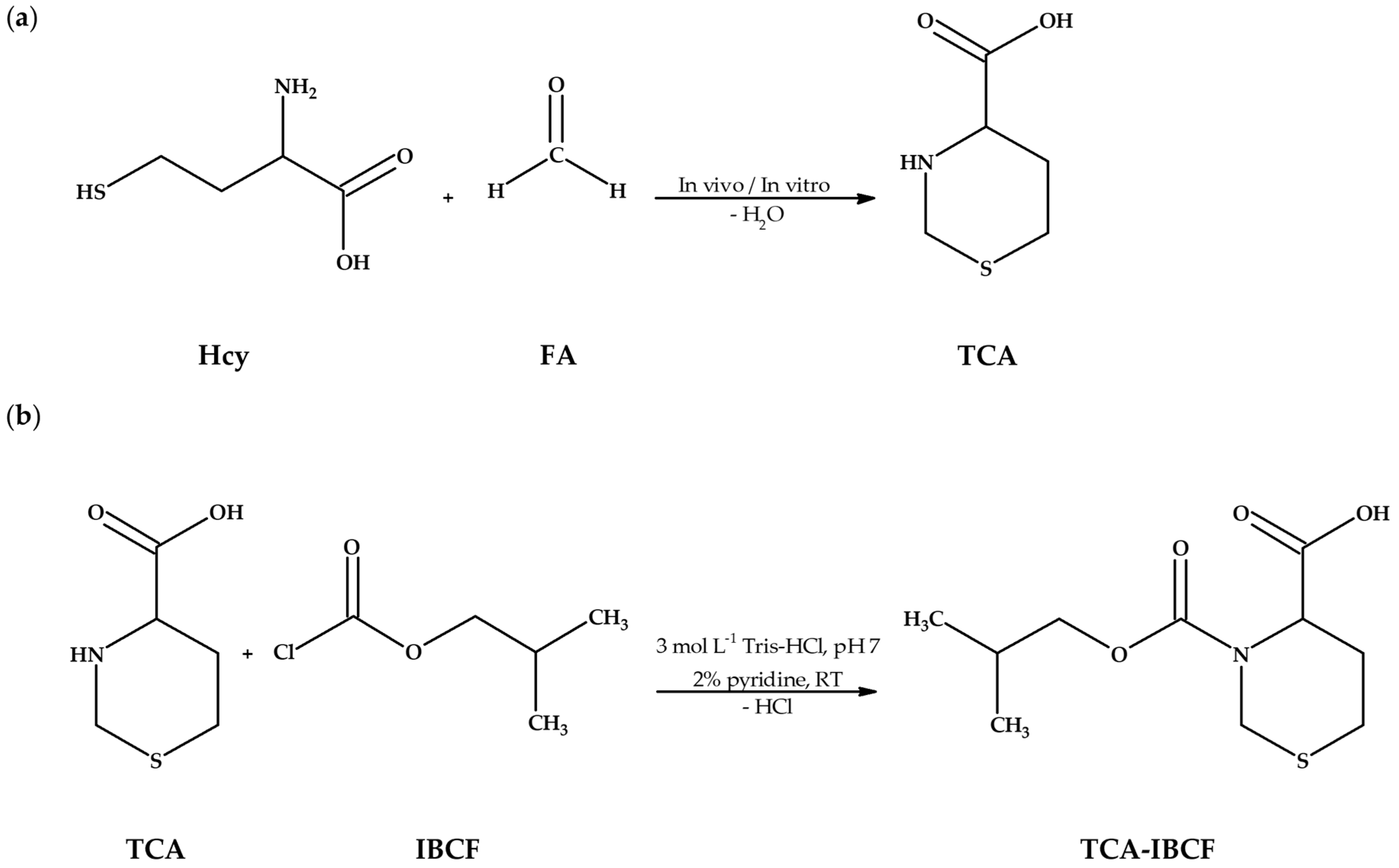
Research project objectives. (a) The reaction equation of homocysteine (Hcy) with formaldehyde (FA) providing 1,3-thiazinane-4-carboxylic acid (TCA); (b) The chemical derivatization reaction of TCA with isobutyl chloroformate (IBCF) in the presence of pyridine, resulting in formation of an isobutyl derivative of TCA (TCA-IBCF). Abbreviations: FA, formaldehyde; Hcy, homocysteine; IBCF, isobutyl chloroformate; RT, room temperature; TCA, 1,3-thiazinane-4-carboxylic acid; TCA-IBCF, isobutyl derivative of 1,3-thiazinane-4-carboxylic acid; Tris-HCl, tris(hydroxymethyl)aminomethane-hydrochloride acid buffer.
Figure 1.
Research project objectives. (a) The reaction equation of homocysteine (Hcy) with formaldehyde (FA) providing 1,3-thiazinane-4-carboxylic acid (TCA); (b) The chemical derivatization reaction of TCA with isobutyl chloroformate (IBCF) in the presence of pyridine, resulting in formation of an isobutyl derivative of TCA (TCA-IBCF). Abbreviations: FA, formaldehyde; Hcy, homocysteine; IBCF, isobutyl chloroformate; RT, room temperature; TCA, 1,3-thiazinane-4-carboxylic acid; TCA-IBCF, isobutyl derivative of 1,3-thiazinane-4-carboxylic acid; Tris-HCl, tris(hydroxymethyl)aminomethane-hydrochloride acid buffer.
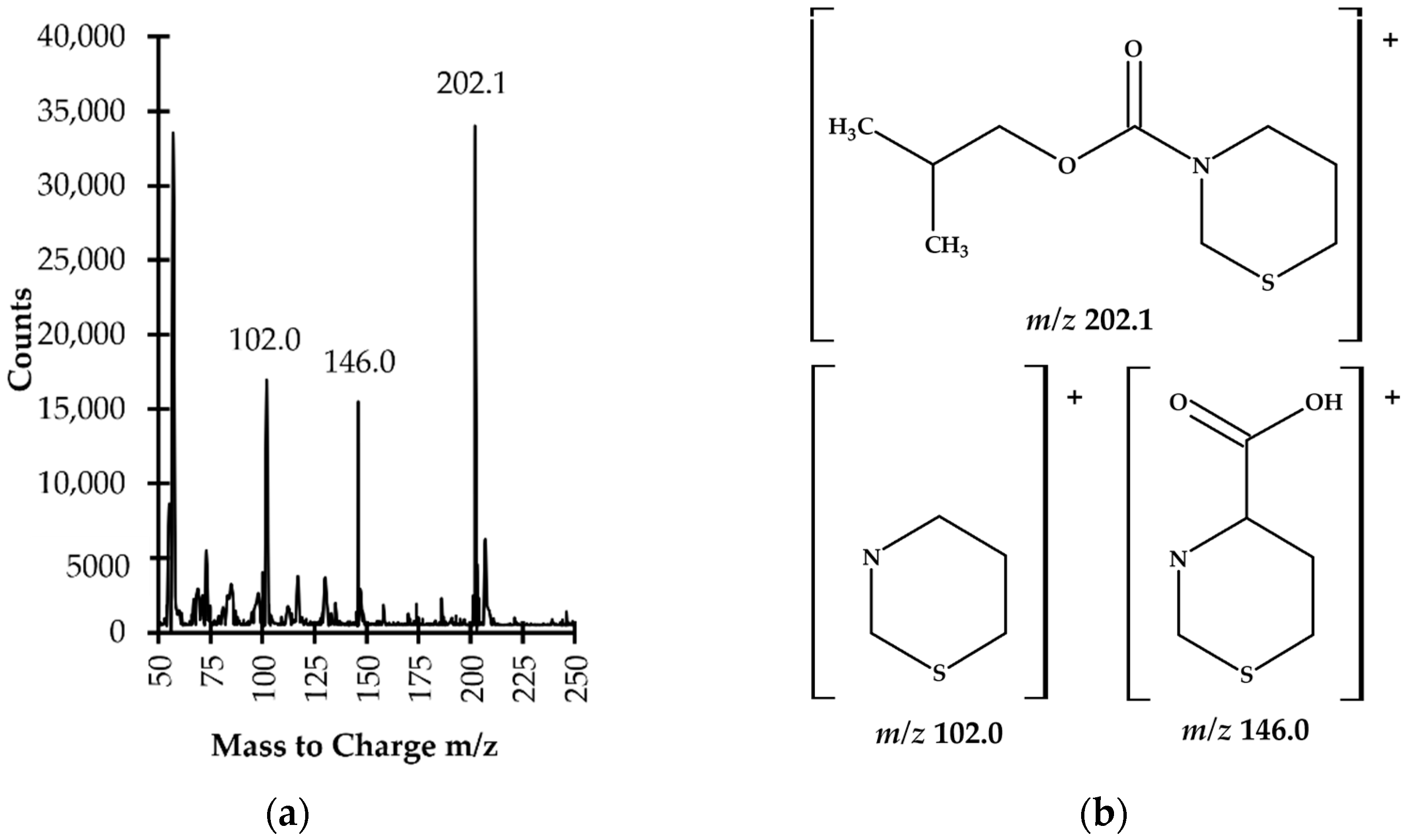
Firstly, it was shown that upon derivatization TCA was converted into its corresponding isobutyl derivative, namely TCA-IBCF which produced intense specific fragment ion peaks, suitable for the analyte monitoring (see
Section 2.2, Figure 6). In addition, it was found that chemical modification was also valuable for increasing the molecular weight of the analyte which helped separate the GC sample peaks from the solvent front. TCA possessed two potential places accessible to IBCF, provided by the amine functional group and carboxyl group [
24]. However, fragmentation ions, found during the study, indicated that the carboxyl group in the molecule remained unmodified under set conditions (see
Section 3.5). Importantly, such circumstances did not affect the effective separation and detection of TCA. In relation to TCA, the IBCF-mediated derivatization generally involved the replacement of the active hydrogen on the amine group with the isobutyl formate moiety, followed by hydrochloric acid release under pyridine catalysis, yielding less polar, more volatile and a relatively thermally stable TCA-IBCF derivative. Thus, the most probable schematic of the derivatization reaction of TCA with IBCF is shown in
Figure 1b.
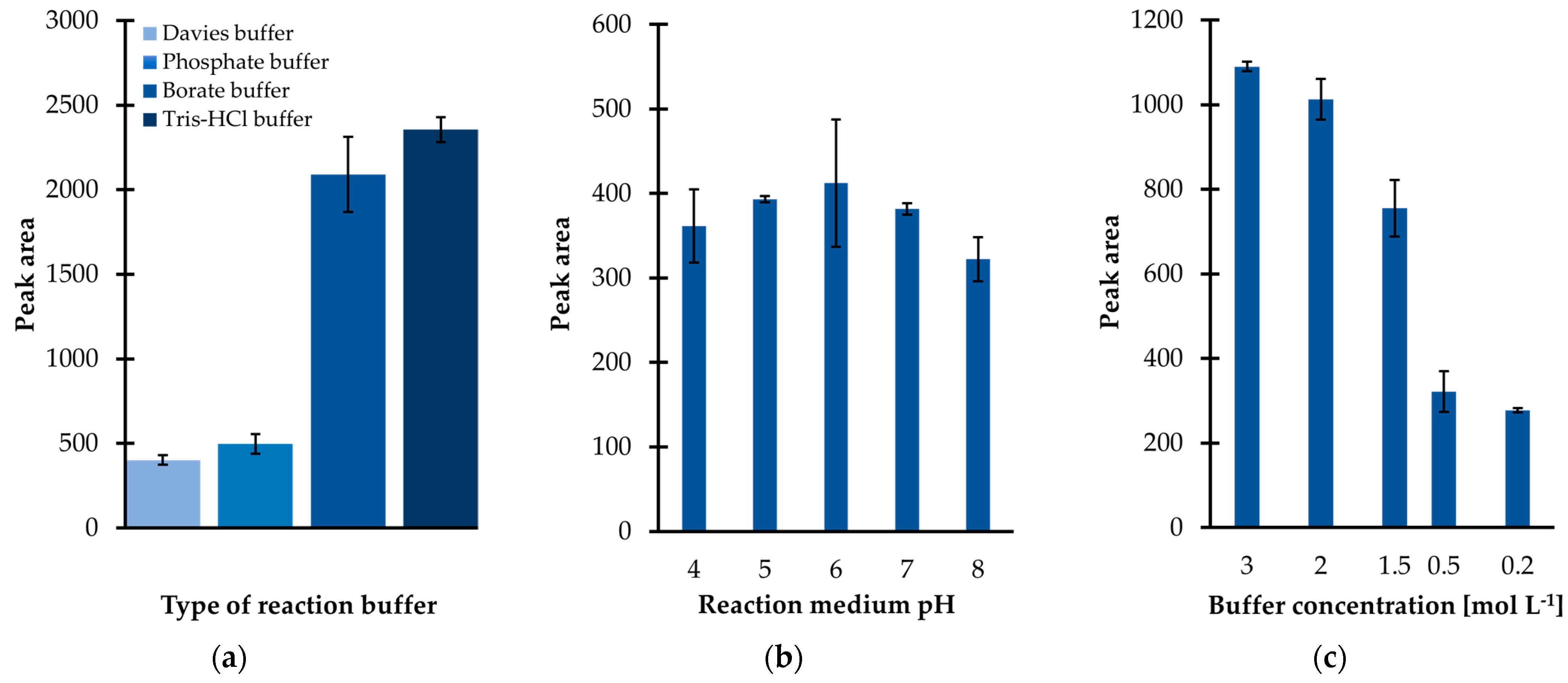
Since the preliminary research results were promising, subsequently the influence of several factors on derivatization efficiency were carefully studied. Firstly, it was recognized that the yield of the IBCF-mediated acylation was highly dependent on the composition of the reaction medium. Several buffer solutions, including phosphate (PB), borate, tris(hydroxymethyl)aminomethane-hydrochloride (Tris-HCl), and Davies buffer were tested. Among them, the Tris-HCl buffer was found to be superior with regard to derivatization and extraction efficiency since the signal intensity of the TCA-IBCF derivative was greater than that registered when other buffer solutions, at the given pH and total buffer concentration, were used (
Figure 2a). As a result, Tris-HCl was chosen to adjust the sample pH regardless of the fact that it contained functional groups, namely hydroxyl and amino groups, that might react with the derivatizing agent of choice [
24]. As shown in Figure 5 (see
Section 2.2), no interference at the retention time of the analyte originating from Tris-HCl buffer was registered, indicating that the particular buffer was suitable as the derivatization reaction medium.
In parallel, the influence of the pH of the reaction medium on derivatization efficiency was evaluated, as the molecule of TCA can exist in different ionization states, which may have an impact on its reactivity. In addition, it was recognized that the reaction medium pH may not only have an effect on the analyte’s reactivity but also on the efficiency of the subsequent sample preparation step, namely extraction. In the presented study, the effect of pH within the range of 4 to 8 was thus evaluated, corresponding to the expected pH of urine under normal conditions. An universal buffer solution of Davies, which has been demonstrated to be suitable for use over the pH range 2 to 12 [
25], was utilized to control the pH level. When analyzing
Figure 2b, a moderate pH dependence on process efficiency can be noticed, suggesting that slightly acidic or even neutral conditions are essential to provide the best results. However, a pH lower than 4 or greater than 7, for the solution, may have a negative effect on the derivatization efficiency as the peak area of the TCA-IBCF derivative decreased with decreasing/increasing pH, respectively (
Figure 2b). In this way, the limited stability of the obtained derivative in strongly acidic/alkaline aqueous media must be considered. Since the useful pH range of the preselected buffering medium is 7 to 9, Tris-HCl buffer, with a pH of 7 was chosen for further experiments.
Next, experiments were conducted in order to optimize the ionic strength of the buffer solution, which was the next factor affecting the particular processes yield. Different Tris-HCl buffer solutions were prepared at five concentration levels, ranging from 0.2to 3 mol L
−1. As shown in
Figure 2c, a progressive increase in the peak area of the TCA-IBCF derivative was observed in parallel with the rise in concentration of Tris-HCl from 0.2 to 3 mol L
−1. In addition, the improvement of the precision of the experiment was concomitantly noticed. As a result, a further 3 mol L
−1 solution of the buffer was used.
Additional experimental work was undertaken to establish the optimal ratio of urine to 3 mol L−1 Tris-HCl buffer, pH 7. Taking into account that the pH of the reaction medium affected the chemical derivatization and extraction processes yield, as well as the fact that urine pH can range from 4 to 8 under normal conditions according to the American Association for Clinical Chemistry, an adjustment to the pH of the particular sample was necessary to minimize its contribution to the recovery losses in this method. In order to evaluate the optimal ratio of urine to the buffer solution of choice, a series of three measurements were performed using pooled urine samples, made up of small pools of the specimens from all donations, that were produced in our laboratory for this purpose. Firstly, the pH of the particular sample was adjusted to a specific pH within the pH range of 4 to 8 by adding a few drops of either diluted hydrochloric acid or sodium hydroxide solution to the sample until the desired pH was achieved. Next, experiments were performed to establish the optimal amount of the buffer by mixing urine samples with different volumes of 3 mol L−1 Tris-HCl buffer, pH 7. Finally, it was established that a minimum ratio of 1:4, urine to buffer was necessary to achieve a pH for the mixture at the same level as the pH of the buffer added to the sample, regardless of the initial pH of the particular sample or its source. As described in the following sections of the paper, the most satisfactory results were obtained when 50 µL of urine were mixed with 200 µL of 3 mol L−1 Tris-HCl buffer, pH 7.
Figure 2.
Derivatization reaction yield as a function of (
a) the type of derivatization buffer; (
b) pH of the reaction medium; (
c) tris(hydroxymethyl)aminomethane-hydrochloride buffer (Tris-HCl) pH 7 concentration, expressed as the peak area of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF). Samples were analyzed according to the procedure described in
Section 3.6. Error bars refer to the standard deviation (SD) of the data (
n = 3). Abbreviations: Tris-HCl, tris(hydroxymethyl)aminomethane-hydrochloride acid buffer.
Figure 2.
Derivatization reaction yield as a function of (
a) the type of derivatization buffer; (
b) pH of the reaction medium; (
c) tris(hydroxymethyl)aminomethane-hydrochloride buffer (Tris-HCl) pH 7 concentration, expressed as the peak area of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF). Samples were analyzed according to the procedure described in
Section 3.6. Error bars refer to the standard deviation (SD) of the data (
n = 3). Abbreviations: Tris-HCl, tris(hydroxymethyl)aminomethane-hydrochloride acid buffer.
According to the literature [
24], alkyl chloroformates are not only influenced by the solvent system, but also by the addition of a catalyst and optional alcohol. In particular, a basic catalyst acting as an acid scavenger is required to neutralize hydrochloric acid, liberated by the derivatization process, in order to drive the reaction forward and/or increase the reactivity of the reagent itself. In parallel, the presence of an auxiliary agent, namely alcohol, in the reaction medium is not prerequisite although it could easily enhance the derivatization yield by means of dispersing the sedimental (cloudy) phase which appears as a result of IBCF and catalyst addition.
In the presented study, one of the most commonly used catalysts, namely pyridine, was employed to facilitate the IBCF assisted reactions. Since pyridine is considered to be toxic, preliminary experiments were conducted to check whether its presence was indispensable for the derivatization. Two sets of samples were prepared; one was treated with IBCF and pyridine, the other with the derivatizing agent itself. Importantly, in the absence of pyridine the outcome of the reaction was rather disappointing. These studies thus have confirmed that the transformation of TCA to the
N-acylated product with IBCF is promoted by the use of pyridine as an acid-binding agent [
24]. Therefore, the influence of the concentration of the catalyst of choice in the study samples on derivatization efficiency was evaluated afterwards. In particular, the effect of the pyridine concentration within the range 0.06 to 0.37 mol L
−1 was tested, corresponding to the addition of 1.5–9 µL of pyridine per 300 µL of reaction mixture. It was found that the derivatization efficiency was enhanced in parallel with an increase in concentration of the catalyst from 0.06 to 0.25 mol L
−1 (0.5–2%), and then the peak area of the TCA-IBCF derivative decreased with the further increasing its concentration. Interestingly, the phenomenon of progressive alkyl chloroformate decomposition along with the diminished efficiency of the derivatization process in the presence of excess pyridine seemed to be characteristic for the course of the reaction [
24]. As a result, 6 µL of pyridine was found to be optimal and was taken for further experiments. Moreover, it is worth mentioning that higher volumes of the catalyst were excluded as they gave rise to excessive baseline noise.
Subsequently, additional experiments were undertaken to determine whether the presence of alcohol was essential for the derivatization reaction or had a negligible effect on the course of the reaction. Two sets of samples were prepared; the samples were treated with IBCF and pyridine in the presence or absence of the corresponding alkyl alcohol. Importantly, the same process yield was achieved even without alcohol in the reaction medium. For this reason, we decided not to use a dispersant in order to reduce chemical consumption, and instead, we chose to shake the sample to achieve dispersion. Indeed, it was recognized that gently shaking samples by hand was sufficient to effectively modify the analyte. In addition, it needs to be emphasized that when samples were shaken violently, vigorously stirred/vortexed or placed in an ultrasonic bath, it resulted in an explosion and the subsequent loss of the sample.
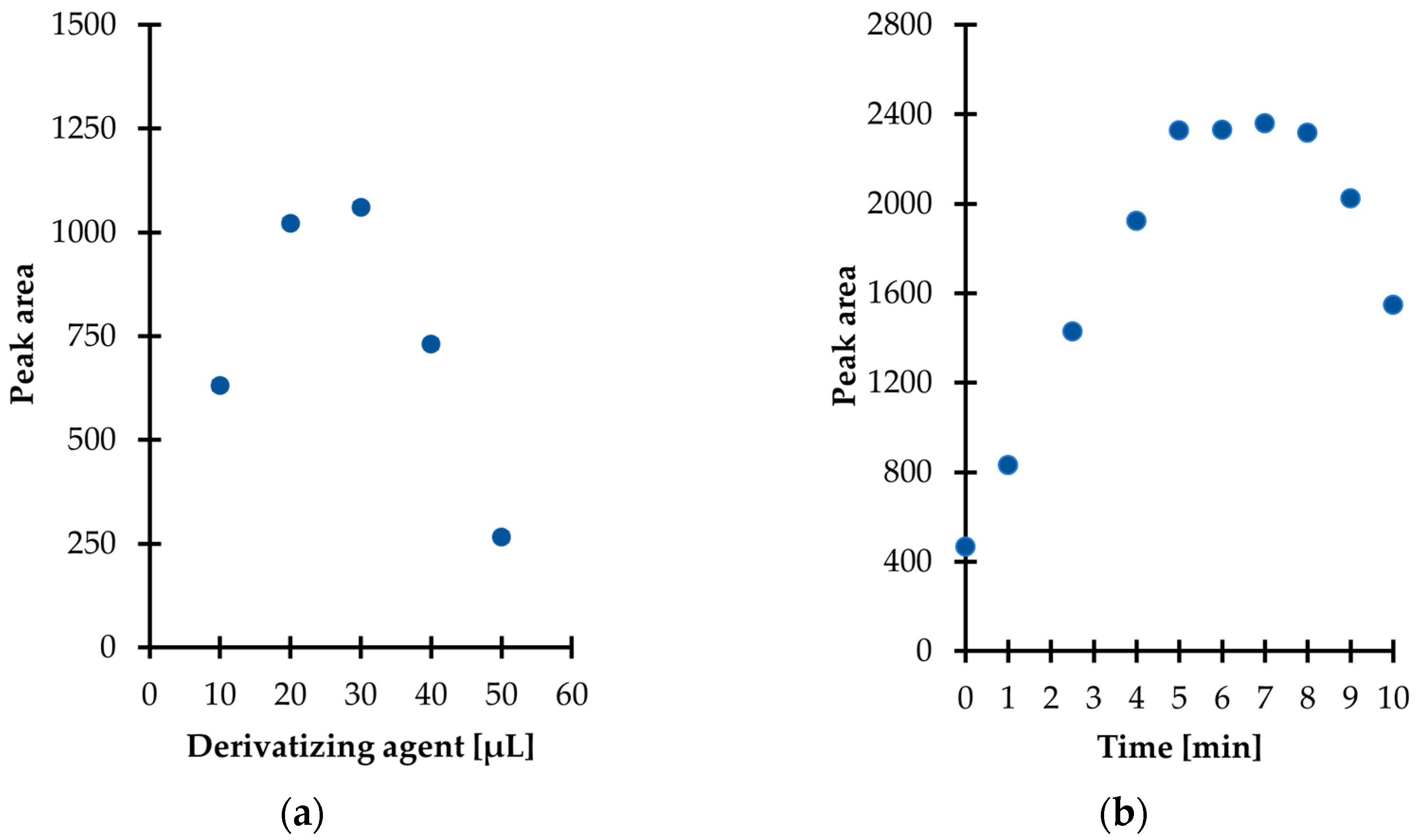
Then, experiments were performed to establish the optimal amount of derivatization reagent for TCA derivatization in urine. Five different quantities, namely 10, 20, 30, 40, and 50 µL were tested, providing the concentration of IBCF in the study samples in the range of 0.27–1.34 mol L
−1. As shown in
Figure 3a, the derivatization efficiency was enhanced in parallel with an increase in the of IBCF from 0.27 to 0.81 mol L
−1, and then the trend towards higher yield was reversed with further increasing its content in the reaction mixture. Importantly, this phenomenon was not surprising as it has been reported elsewhere that the reaction products may be decomposed when excessive amounts of IBCF over pyridine is used [
24]. Therefore, this fact cannot be neglected in any attempt to employ IBCF and other alkyl chloroformates as derivatizing agents as the presence of their excess may result in the occurrence of side reactions along with diminished efficiency of the derivatization process. Finally, 30 µL of IBCF was taken for further experiments as it gave rise to the best results (
Figure 3a). Importantly, it has been recognized that pyridine and IBCF have to be added to the reaction mixture separately, since mixing them beforehand results in a cloudy solution which impedes precise pipetting.
Further experiments were conducted in order to establish the derivatization reaction kinetics at room temperature. In parallel, the stability of the obtained derivative in aqueous media was also evaluated. In particular, it was established that the reaction at the above-defined conditions was completed in just 5 min after mixing the reagents (
Figure 3b). Therefore, no experimental work was undertaken to further optimize the temperature of the reaction. Notably, it was also found that the TCA-IBCF derivative signal remained stable no longer than 3 min under set conditions, as a progressive decrease of the signal peak area was observed (
Figure 3b). Hence, it is highly recommended to subject samples to the subsequent processing step, namely extraction just after completion of the derivatization reaction. It means that the extraction solvent should be added to the reaction mixture within 5 min and no later than 8 min after mixing the reagents together; this was considered well within the sample stability and derivatization efficiency, as well.
In summary, our research led us to the conclusion that Tris-HCl buffer with pyridine proved to be a suitable derivatization reaction medium, while the mere addition of IBCF led to the almost immediate formation of the desirable reaction product. As a result, for routine analysis, 50 µL of urine was initially mixed with 200 µL 3 mol L
−1 Tris-HCl buffer, pH 7 and was then treated with 6 µL pyridine and 30 µL IBCF, resulting in the final concentration in urine samples of 0.26 mol L
−1 (~2.1%) and 0.81 mol L
−1 (~10.5%), respectively. As described above, processing samples for 5 min at room temperature, after gentle shaking by hand, was sufficient to effectively modify the analyte. Importantly, such an approach was also advantageous as it provided good repeatability of the reaction. From a purely practical point of view, an explosion and subsequent loss of the sample was not observed under these conditions. Notably, analyte dilution was not a limitation as samples were concentrated in the subsequent processing step (see
Section 2.1.2).
Figure 3.
Derivatization reaction yield as a function of (
a) the amount of the reagent; (
b) time combined with examination of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF) stability in the aqueous phase, expressed as a peak area of the derivative. Samples were analyzed according to the procedure described in
Section 3.6.
Figure 3.
Derivatization reaction yield as a function of (
a) the amount of the reagent; (
b) time combined with examination of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF) stability in the aqueous phase, expressed as a peak area of the derivative. Samples were analyzed according to the procedure described in
Section 3.6.
2.1.2. Sample Purification
It has long been known that water-containing samples cannot be directly injected into a GC–MS system without purification prior to analysis. According to the literature, a number of chemical and physical purification methods have been identified, and the most commonly used techniques to adapt sample matrices to be more compatible with the target chromatographic method involve extraction [
26].
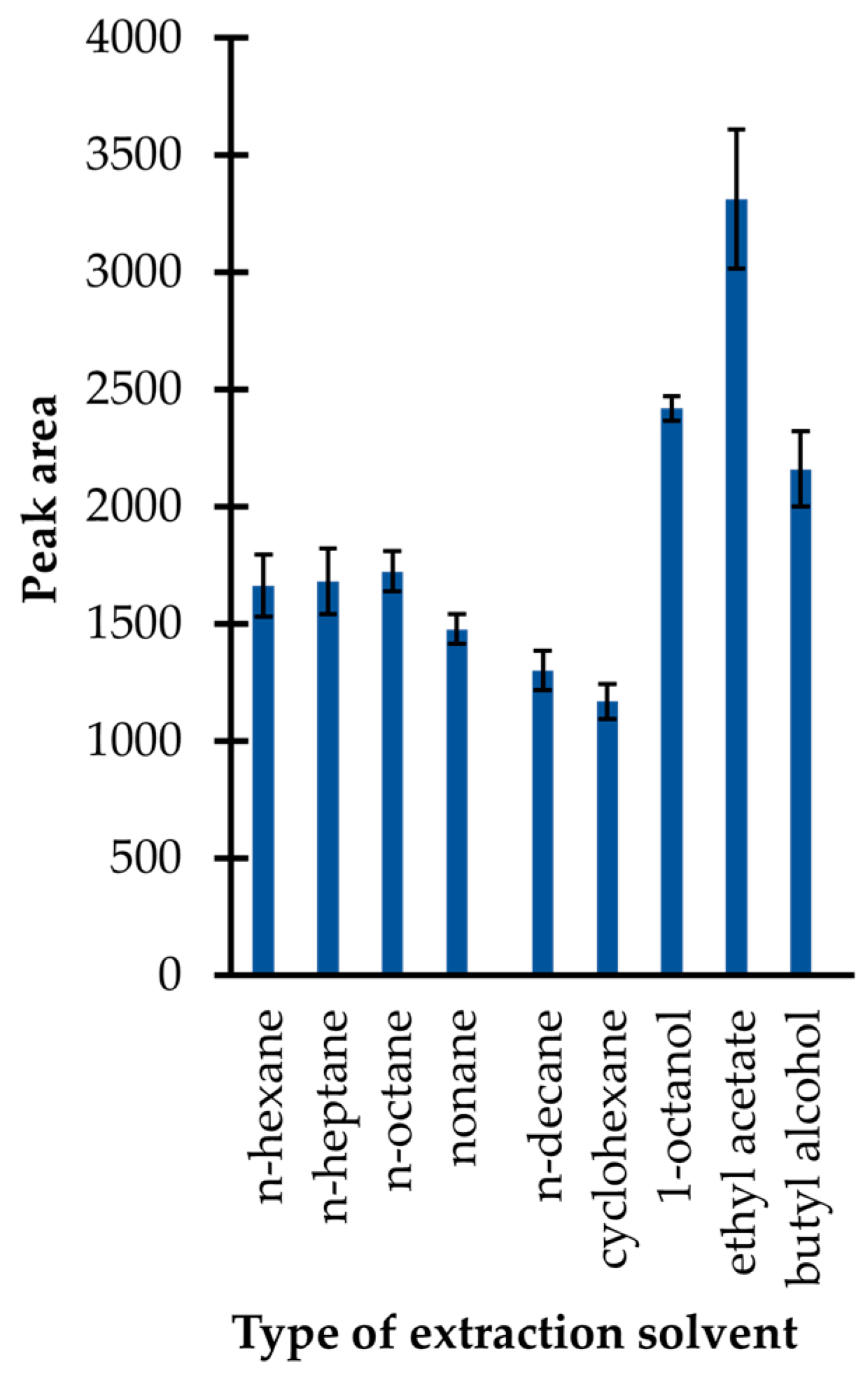
In the presented study, an approach utilizing simple liquid–liquid extraction was used. Firstly, several immiscible solvents with lower density than water, including n-hexane, n-heptane, n-octane, nonane, n-decane, cyclohexane, ethyl acetate, 1-octanol, and butyl alcohol were tested. In general, the selection of extractants was made on the basis of findings that the upper organic (acceptor) phase is easier to collect for a GC–MS analysis than the bottom one. As shown in
Figure 4, the application of ethyl acetate resulted in the highest extraction efficiency, among other preselected organic solvents. In addition, it was identified that its application resulted in less complex chromatograms in comparison with those registered when other organic solvents were used. Therefore, ethyl acetate was selected.
Then, experiments were performed to establish the optimal amount of the organic solvent of choice. Six different volumes, namely 50, 100, 150, 200, 250 and 300 µL, were tested that amounted to 1 to 6-times the volume of the urine specimen. In particular, it was found that the extraction efficiency decreased with the increase in the extractant volume, due to a substantial dilution of the sample (analyte). Indeed, an efficient extraction was achieved by mixing the resulting mixture with just 50 µL of ethyl acetate, while a minimum volume of 100 µL extractant was found to be necessary to provide both efficient extraction, and sufficiently large enough volume of acceptor phase to enable easy collection for the GC–MS analysis. Thus, 100 µL of ethyl acetate was selected as it was considered satisfactory for ensuring sample handling convenience.
Further experiments were conducted to obtain information about the extraction conditions that promoted the process. In parallel, the stability of the TCA-IBCF derivative in organic media was examined. In particular, it was established that shaking/vortexing the samples followed by centrifugation did not markedly affect operational efficiency, which was beneficial for workflow simplification. Nevertheless, centrifugation was pointed out to be a necessary part of the sample preparation process as signals were higher by about 60% under the discussed conditions in comparison with those registered when the extraction was not accelerated by spinning the samples in solution around an axis at high speed. Notably, it was also found that the extraction under the above-defined conditions was completed in just 1 min after mixing the reagents and immediately placing the samples in the centrifuge. In parallel, it was established that the TCA-IBCF derivative signal remained stable no longer than subsequent 4 min under the set conditions, and then the extraction efficiency decreased dramatically with the prolonged processing time, which was tested in the range of 5–10 min. Equally importantly, the same observations were made when the stability of the study samples in the autosampler, prepared according to the procedure described in
Section 3.5, were evaluated. As a result, we were able to conclude from this study that the obtained derivative is fairly unstable under these set conditions, indicating the necessity of subjecting the samples to the GC–MS analysis just after their preparation in order to produce meaningful results. On the other hand, it was important to recognize that the TCA-IBCF derivative did not undergo thermal decomposition in the GC system under optimized conditions (see
Section 3.6) and produced a single symmetrical peak (see
Section 2.2,
Figure 5), although high thermal processing temperatures during GC–MS analysis may potentially lead to the formation of degradation products [
27]. Finally, for routine analysis, 286 µL of the resulting mixture and 100 µL of ethyl acetate were mixed together, and then placed in the centrifuge at 6000 rpm for 1 min at room temperature. Under these conditions, the cloudy (upper) organic phase was obtained, which was then collected and injected into the GC–MS system immediately.
Figure 4.
Extraction efficiency of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF) as a function of the type of extraction solvent expressed as a peak area of the derivative. Samples were analyzed according to the procedure described in
Section 3.6. Error bars refer to the standard deviation (SD) of the data (
n = 3).
Figure 4.
Extraction efficiency of the isobutyl derivative of 1,3-thiazinane-4-carboxylic acid (TCA-IBCF) as a function of the type of extraction solvent expressed as a peak area of the derivative. Samples were analyzed according to the procedure described in
Section 3.6. Error bars refer to the standard deviation (SD) of the data (
n = 3).
In summary, these experiments established an optimal procedure, in which urine was treated with a derivatizing agent, namely IBCF in the presence of pyridine, and subjected to the GC–MS system without delay after ethyl acetate extraction. Importantly, this is a simple one-pot sample processing method for urinary TCA determination, in which the overall sample preparation time was estimated to be 10 min, taking into account all the operations that need to be performed, including derivatization, extraction, centrifugation, and pipetting etc. Furthermore, the process was accompanied by consumption of 336 µL of inexpensive chemicals. In our opinion, the GC–MS assay can thus be considered environmentally-friendly thanks to the possibility of carrying out the chemical analysis on a very small scale combined with low consumption of hazardous chemicals and laboratory disposable plasticware. Nevertheless, it needs to be emphasized that a successful analysis using the proposed method can only be achieved when the recommended sample handling and management procedures, described herein, are followed.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}