
Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling


Abstract
:1. Introduction
2. Problems and Prospects of iPSCs-Based Models of Neurodegenerative Diseases
3. Obtaining the iPSCs-Based Models of Neurodegenerative Diseases
4. Neurodegeneration and Calcium Signaling
5. Alzheimer’s Disease
6. Parkinson’s Disease
7. Huntington’s Disease
8. Spinocerebellar Ataxias
9. Amyotrophic Lateral Sclerosis
10. Other Neurological Diseases
10.1. Autism Spectrum Disorders
10.2. Leigh’s Syndrome
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Dementia Factsheet. Available online: https://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed on 1 December 2021).
- Ciobanu, A.M.; Ionita, I.; Buleandra, M.; David, I.G.; Popa, D.E.; Ciucu, A.A.; Budisteanu, M. Current advances in metabolomic studies on non-motor psychiatric manifestations of Parkinson’s disease (Review). Exp. Ther. Med. 2021, 22, 1010. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S. Towards theories of brain aging. In Handbook of Studies on Psychiatry and Old Age; Kay, D.S., Burrows, G.W., Eds.; Elsevier: Amsterdam, The Netherlands, 1984; pp. 7–30. [Google Scholar]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, P.; Popugaeva, E.; Bezprozvanny, I. Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin. Cell Dev. Biol. 2015, 40, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.S.; Lin, H.Y.; Lee-Chen, G.J.; Hsieh-Li, H.M.; Wu, C.H.; Lin, J.Y. Treatment with a Ginkgo biloba extract, EGb 761, inhibits excitotoxicity in an animal model of spinocerebellar ataxia type 17. Drug. Des. Dev. Ther. 2016, 10, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeredys, M.; Vigont, V.A.; Boeva, V.A.; Mikoshiba, K.; Kaznacheyeva, E.V.; Kuznicki, J. Huntingtin-Associated Protein 1A Regulates Store-Operated Calcium Entry in Medium Spiny Neurons from Transgenic YAC128 Mice, a Model of Huntington’s Disease. Front. Cell Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, C.; Hamada, K.; Mikoshiba, K. Ca2+ signaling and spinocerebellar ataxia. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865 Pt B, 1733–1744. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Freude, K.K.; Penjwini, M.; Davis, J.L.; LaFerla, F.M.; Blurton-Jones, M. Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J. Biol. Chem. 2011, 286, 24264–24274. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Stüber, K.; Poppe, D.; Doerr, J.; Ladewig, J.; Brüstle, O.; Koch, P. Embryonic stem cell-based modeling of tau pathology in human neurons. Am. J. Pathol. 2013, 182, 1769–1779. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X.; et al. ESC-Derived Basal Forebrain Cholinergic Neurons Ameliorate the Cognitive Symptoms Associated with Alzheimer’s Disease in Mouse Models. Stem Cell Rep. 2015, 5, 776–790. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Chen, J.; Deng, X.; Liu, Y.; Rao, M.S.; Cadet, J.L.; Freed, W.J. An In Vitro model of human dopaminergic neurons derived from embryonic stem cells: MPP+ toxicity and GDNF neuroprotection. Neuropsychopharmacology 2006, 31, 2708–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.L.; Seehus, C.R.; Capowski, E.E.; Aebischer, P.; Zhang, S.C.; Svendsen, C.N. Over-expression of alpha-synuclein in human neural progenitors leads to specific changes in fate and differentiation. Hum. Mol. Genet. 2007, 16, 651–666. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Aubry, L.; Bugi, A.; Lefort, N.; Rousseau, F.; Peschanski, M.; Perrier, A.L. Striatal progenitors derived from human ES cells mature into DARPP32 neurons In Vitro and in quinolinic acid-lesioned rats. Proc. Natl. Acad. Sci. USA 2008, 105, 16707–16712. [Google Scholar] [CrossRef] [Green Version]
- Niclis, J.; Trounson, A.O.; Dottori, M.; Ellisdon, A.; Bottomley, S.P.; Verlinsky, Y.; Cram, D. Human embryonic stem cell models of Huntington disease. Reprod. Biomed. Online 2009, 19, 106–113. [Google Scholar] [CrossRef]
- Moore, L.R.; Keller, L.; Bushart, D.D.; Delatorre, R.G.; Li, D.; McLoughlin, H.S.; do Carmo Costa, M.; Shakkottai, V.G.; Smith, G.D.; Paulson, H.L. Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Res. 2019, 39, 101504. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Karumbayaram, S.; Kelly, T.K.; Paucar, A.A.; Roe, A.J.; Umbach, J.A.; Charles, A.; Goldman, S.A.; Kornblum, H.I.; Wiedau-Pazos, M. Human embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALS. Dis. Models Mech. 2009, 2, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013, 12, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum Mol Genet 2018, 27, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Yool, A.J.; Bill, R.M. Recent breakthroughs and future directions in drugging aquaporins. Trends Pharmacol. Sci. 2022, 43, 30–42. [Google Scholar] [CrossRef]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 ΔE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [Green Version]
- Shaltouki, A.; Sivapatham, R.; Pei, Y.; Gerencser, A.A.; Momčilović, O.; Rao, M.S.; Zeng, X. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 2015, 4, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Kawakami, H.; Kitajima, H.; Nishiyama, A.; Sasai, Y.; Inoue, H.; Muguruma, K. Vulnerability of Purkinje Cells Generated from Spinocerebellar Ataxia Type 6 Patient-Derived iPSCs. Cell Rep. 2016, 17, 1482–1490. [Google Scholar] [CrossRef] [Green Version]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [Green Version]
- Ooi, J.; Langley, S.R.; Xu, X.; Utami, K.H.; Sim, B.; Huang, Y.; Harmston, N.P.; Tay, Y.L.; Ziaei, A.; Zeng, R.; et al. Unbiased profiling of isogenic Huntington disease hPSC-derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell. Rep. 2019, 123, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Tagliafierro, L.; Zamora, M.E.; Chiba-Falek, O. Multiplication of the SNCA locus exacerbates neuronal nuclear aging. Hum. Mol. Genet. 2019, 28, 407–421. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-Organization of Polarized Cerebellar Tissue in 3D Culture of Human Pluripotent Stem Cells. Cell. Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Conforti, P.; Besusso, D.; Bocchi, V.D.; Faedo, A.; Cesana, E.; Rossetti, G.; Ranzani, V.; Svendsen, C.N.; Thompson, L.M.; Toselli, M.; et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E762–E771. [Google Scholar] [CrossRef] [Green Version]
- Świtońska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, Ł.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of altered developmental pathways in human juvenile HD iPSC with 71Q and 109Q using transcriptome profiling. Front Cell Neurosci. 2018, 12, 528. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain. 2012, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef] [Green Version]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by definedfactors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambasudhan, R.; Talantova, M.; Coleman, R.; Yuan, X.; Zhu, S.; Lipton, S.A.; Ding, S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 2011, 9, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Victor, M.B.; Richner, M.; Hermanstyne, T.O.; Ransdell, J.L.; Sobieski, C.; Deng, P.Y.; Klyachko, V.A.; Nerbonne, J.M.; Yoo, A.S. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 2014, 84, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct conversionof normal and Alzheimer’s disease human fibroblasts intoneuronal cells by small molecules. Cell Stem Cell. 2015, 17, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Stummann, T.C.; Borland, H.; Hasholt, L.F.; Tümer, Z.; Nielsen, J.E.; Rasmussen, M.A.; Nielsen, T.T.; Daechsel, J.C.; Fog, K.; et al. Induced pluripotent stem cell—Derived neurons for the study of spinocerebellar ataxia type 3. Stem Cell Res. 2016, 17, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Z.; Luo, M.; Niu, X.; Chen, Y.; Xie, Y.; He, W.; Song, B.; Xian, Y.; Fan, D.; OuYang, S.; et al. Autophagy Promoted the Degradation of Mutant ATXN3 in Neurally Differentiated Spinocerebellar Ataxia-3 Human Induced Pluripotent Stem Cells. Biomed. Res. Int. 2016, 6701793. [Google Scholar] [CrossRef] [Green Version]
- Vigont, V.A.; Grekhnev, D.A.; Lebedeva, O.S.; Gusev, K.O.; Volovikov, E.A.; Skopin, A.Y.; Bogomazova, A.N.; Shuvalova, L.D.; Zubkova, O.A.; Khomyakova, E.A.; et al. STIM2 Mediates Excessive Store-Operated Calcium Entry in Patient-Specific iPSC-Derived Neurons Modeling a Juvenile Form of Huntington’s Disease. Front. Cell Dev. Biol. 2021, 9, 625231. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Qi, Y.; Mica, Y.; Lee, G.; Zhang, X.J.; Niu, L.; Bilsland, J.; Cao, L.; Stevens, E.; Whiting, P.; et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 2012, 30, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Bruijn, L.I.; Chandran, S.; Eggan, K.; Ho, R.; Klim, J.R.; Livesey, M.R.; Lowry, E.; Macklis, J.D.; Rushton, D.; et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 2016, 19, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef]
- Filippini, A.; Mutti, V.; Faustini, G.; Longhena, F.; Ramazzina, I.; Rizzi, F.; Kaganovich, A.; Roosen, D.A.; Landeck, N.; Duffy, M.; et al. Extracellular clusterin limits the uptake of α-synuclein fibrils by murine and human astrocytes. Glia 2021, 69, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess α-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.L.; Cui, Q.L.; Schambach, A.; Kim, K.P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katt, M.E.; Mayo, L.N.; Ellis, S.E.; Mahairaki, V.; Rothstein, J.D.; Cheng, L.; Searson, P.C. The role of mutations associated with familial neurodegenerative disorders on blood-brain barrier function in an iPSC model. Fluids Barriers CNS 2019, 16, 20. [Google Scholar] [CrossRef]
- HD iPSC Consortium. Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci. 2017, 20, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Abuammar, H.; Bhattacharjee, A.; Simon-Vecsei, Z.; Blastyák, A.; Csordás, G.; Páli, T.; Juhász, G. Ion Channels and Pumps in Autophagy: A Reciprocal Relationship. Cells 2021, 10, 3537. [Google Scholar] [CrossRef]
- Kelemen, K.; Szilágyi, T. New Approach for Untangling the Role of Uncommon Calcium-Binding Proteins in the Central Nervous System. Brain Sci. 2021, 11, 634. [Google Scholar] [CrossRef]
- Latoszek, E.; Czeredys, M. Molecular Components of Store-Operated Calcium Channels in the Regulation of Neural Stem Cell Physiology, Neurogenesis, and the Pathology of Huntington’s Disease. Front Cell. Dev. Biol. 2021, 9, 657337. [Google Scholar] [CrossRef]
- Hajieva, P.; Baeken, M.W.; Moosmann, B. The role of Plasma Membrane Calcium ATPases (PMCAs) in neurodegenerative disorders. Neurosci. Lett. 2018, 663, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Van Remmen, H. The SarcoEndoplasmic Reticulum Calcium ATPase (SERCA) pump: A potential target for intervention in aging and skeletal muscle pathologies. Skelet Muscle 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; de Rosa, V.; Cammarota, M.; Secondo, A.; Pannaccione, A.; Annunziato, L. The Na(+)/Ca(2+) exchangers in demyelinating diseases. Cell Calcium 2020, 85, 102130. [Google Scholar] [CrossRef]
- Pannaccione, A.; Piccialli, I.; Secondo, A.; Ciccone, R.; Molinaro, P.; Boscia, F.; Annunziato, L. The Na(+)/Ca(2+)exchanger in Alzheimer’s disease. Cell Calcium 2020, 87, 102190. [Google Scholar] [CrossRef]
- Guan, P.P.; Cao, L.L.; Yang, Y.; Wang, P. Calcium Ions Aggravate Alzheimer’s Disease Through the Aberrant Activation of Neuronal Networks, Leading to Synaptic and Cognitive Deficits. Front. Mol. Neurosci. 2021, 14, 757515. [Google Scholar] [CrossRef]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Hayden, M.R. Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 2004, 322, 1310–1317. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium hypothesis of neurodegeneration—An update. Biochem. Biophys. Res. Commun. 2019, 520, 667–669. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Hoogendijk, W.J.G.; Pool, C.W.; Troost, D.; Van Zwieten, E.; Swaab, D.F. Image analyser-assisted morphometry of the locus coeruleus in alzheimer’s disease, parkinson’s disease and amyotrophic lateral sclerosis. Brain 1995, 118, 131–143. [Google Scholar] [CrossRef]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef]
- Theofilas, P.; Ehrenberg, A.J.; Dunlop, S.; Di Lorenzo Alho, A.T.; Nguy, A.; Leite, R.E.P.; Rodriguez, R.D.; Mejia, M.B.; Suemoto, C.K.; Ferretti-Rebustini, R.E.L.; et al. Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimer’s Dement. 2017, 13, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Chiou, S.J.; Wong, Y.H.; Wang, Y.H.; Lai, Y.; Chou, C.H.; Wang, C.; Loh, J.K.; Lieu, A.S.; Cheng, J.T.; et al. GSKIP-Mediated Anchoring Increases Phosphorylation of Tau by PKA but Not by GSK3beta viacAMP/PKA/GSKIP/GSK3/Tau Axis Signaling in Cerebrospinal Fluid and iPS Cells in Alzheimer Disease. J. Clin. Med. 2019, 8, 1751. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Van Dongen, A.M. Enhanced Neuronal Activity and Asynchronous Calcium Transients Revealed in a 3D Organoid Model of Alzheimer’s Disease. ACS Biomater. Sci. Eng. 2021, 7, 254–264. [Google Scholar] [CrossRef]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based compound screening and In Vitro trials identify a synergistic anti-amyloid beta combination for Alzheimer’s disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Chen, H.; Cross, A.C.; Thakkar, A.; Xu, H.; Li, A.; Paull, D.; Noggle, S.A.; Kruger, L.; Denton, T.T.; Gibson, G.E. Selective linkage of mitochondrial enzymes to intracellular calcium stores differs between human-induced pluripotent stem cells, neural stem cells, and neurons. J. Neurochem. 2021, 156, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, S.S.; Engel, M.; Balez, R.; Do-Ha, D.; Cabral-da-Silva, M.C.; Hernández, D.; Berg, T.; Fifita, J.A.; Grima, N.; Yang, S.; et al. A Simple Differentiation Protocol for Generation of Induced Pluripotent Stem Cell-Derived Basal Forebrain-Like Cholinergic Neurons for Alzheimer’s Disease and Frontotemporal Dementia Disease Modeling. Cells 2020, 9, 2018. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 mutant iPSC-Derived Model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raut, S.; Patel, R.; Al-Ahmad, A.J. Presence of a mutation in PSEN1 or PSEN2 gene is associated with an impaired brain endothelial cell phenotype In Vitro. Fluids Barriers CNS 2021, 18, 3. [Google Scholar] [CrossRef]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 2018, 7, e40070. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Tornroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021, awab311. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Iliff, J.J.; Bill, R.M. Aquaporin 4 and glymphatic flow have central roles in brain fluid homeostasis. Nat. Rev. Neurosci. 2021, 22, 650–651. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Langston, J.W. The Parkinson’s complex: Parkinsonism is just the tip of the iceberg. Ann Neurol. 2006, 59, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013, 13, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; Laganière, J.; Cooper, O.; Mak, S.K.; Vu, B.J.; Huang, Y.A.; Paschon, D.E.; Vangipuram, M.; Sundararajan, R.; Urnov, F.D.; et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol. Dis. 2014, 62, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.J.; Ebert, A.D. Neurite Aggregation and Calcium Dysfunction iniPSC-Derived Sensory Neurons with Parkinson’s Disease-Related LRRK2 G2019SMutation. Stem Cell Rep. 2015, 5, 1039–1052. [Google Scholar] [CrossRef] [Green Version]
- Borgs, L.; Peyre, E.; Alix, P.; Hanon, K.; Grobarczyk, B.; Godin, J.D.; Purnelle, A.; Krusy, N.; Maquet, P.; Lefebvre, P.; et al. Dopaminergic neurons differentiating from LRRK2 G2019S induced pluripotent stem cells show early neuritic branching defects. Sci. Rep. 2016, 6, 33377. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell. 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Neely, M.D.; Davison, C.A.; Aschner, M.; Bowman, A.B. From the Cover: Manganese and Rotenone-Induced Oxidative Stress Signatures Differ in iPSC-Derived Human Dopamine Neurons. Toxicol. Sci. 2017, 159, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [Green Version]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.; et al. Defects in mRNATranslation in LRRK2-Mutant hiPSC-Derived Dopaminergic Neurons Lead toDysregulated Calcium Homeostasis. Cell Stem Cell 2020, 27, 633–645.e7. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Korecka, J.A.; Talbot, S.; Osborn, T.M.; de Leeuw, S.M.; Levy, S.A.; Ferrari, E.J.; Moskites, A.; Atkinson, E.; Jodelka, F.M.; Hinrich, A.J.; et al. Neurite Collapse and Altered ER Ca2+ Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem Cell Rep. 2019, 12, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033. [Google Scholar] [CrossRef] [Green Version]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type calcium channels determine the vulnerability of dopaminergic neurons to mitochondrial stress in familial Parkinson disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Benkert, J.; Hess, S.; Roy, S.; Beccano-Kelly, D.; Wiederspohn, N.; Duda, J.; Simons, C.; Patil, K.; Gaifullina, A.; Mannal, N.; et al. Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson’s disease. Nat. Commun. 2019, 10, 5094. [Google Scholar] [CrossRef]
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and α-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 237. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox. Signal 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Czeredys, M. Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington’s Disease. Front Cell Dev. Biol. 2020, 8, 611735. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Di Figlia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef]
- HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096.e6. [Google Scholar] [CrossRef] [Green Version]
- Camnasio, S.; Delli Carri, A.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Lo Riso, P.; Castiglioni, V.; Zuccato, C.; et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, E.D.; Kiselev, S.L. Mitochondrial distribution violation and nuclear indentations in neurons differentiated from iPSCs of Huntington’s disease patients. J. Stem Cells Regen. Med. 2018, 14, 80–85. [Google Scholar] [CrossRef]
- Vigont, V.; Nekrasov, E.; Shalygin, A.; Gusev, K.; Klushnikov, S.; Illarioshkin, S.; Lagarkova, M.; Kiselev, S.L.; Kaznacheyeva, E. Patient-Specific iPSC-Based Models of Huntington’s Disease as a Tool to Study Store-Operated Calcium Entry Drug Targeting. Front. Pharmacol. 2018, 9, 696. [Google Scholar] [CrossRef]
- Mathkar, P.P.; Suresh, D.; Dunn, J.; Tom, C.M.; Mattis, V.B. Characterization of Neurodevelopmental Abnormalities in iPSC-Derived Striatal Cultures from Patients with Huntington’s Disease. J. Huntingt. Dis. 2019, 8, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington’s Disease Patient-Derived Astrocytes Display Electrophysiological Impairments and Reduced Neuronal Support. Front. Neurosci. 2019, 13, 669. [Google Scholar] [CrossRef] [PubMed]
- HD iPSC Consortium. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet. 2020, 29, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington’s Disease Mouse Model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; den Dunnen, W.; Korf, H.W.; Rüb, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Ono, Y.; Miyawaki, H.; Mizuhara, E.; Hori, S.; Kakizuka, A.; Obata, K.; Yanagawa, Y.; Hirano, T.; et al. Ontogeny-recapitulating generation and tissue integration of ES cell-derived Purkinje cells. Nat. Neurosci. 2010, 13, 1171–1180. [Google Scholar] [CrossRef]
- Muguruma, K.; Sasai, Y. In vitro recapitulation of neural development using embryonic stem cells: From neurogenesis to histogenesis. Dev. Growth Differ. 2012, 54, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Morino, H.; Matsuda, Y.; Muguruma, K.; Miyamoto, R.; Ohsawa, R.; Ohtake, T.; Watanabe, M.; Maruyama, H.; Hashimoto, K.; Kawakami, H.; et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 2015, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, N.; Sora, I.; Muguruma, K. Self-organizing cortex generated from human iPSCs with combination of FGF2 and ambient oxygen. Biochem. Biophys. Res. Commun. 2018, 498, 729–735. [Google Scholar] [CrossRef]
- Tamada, A.; Watanabe, S.; Muguruma, K. Investigating developmental and disease mechanisms of the cerebellum with pluripotent stem cells. Mol. Cell Neurosci. 2020, 107, 103530. [Google Scholar] [CrossRef]
- Wang, S.; Wang, B.; Pan, N.; Fu, L.; Wang, C.; Song, G.; An, J.; Liu, Z.; Zhu, W.; Guan, Y.; et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci. Rep. 2015, 5, 9232. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum 2018, 17, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, D.E.; Carroll, T.S.; Kocabas, A.; Zhu, X.; Behesti, H.; Faust, P.L.; Stalbow, L.; Fang, Y.; Hatten, M.E. Novel genetic features of human and mouse Purkinje cell differentiation defined by comparative transcriptomics. Proc. Natl. Acad. Sci. USA 2020, 117, 15085–15095. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, M.W.C.; Tschumperlin, T.; Lu, H.C.; Lackey, E.P.; Bondar, V.V.; Wan, Y.W.; Tan, Q.; Adamski, C.J.; Friedrich, J.; Twaroski, K.; et al. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 2018, 97, 1235–1243.e1235. [Google Scholar] [CrossRef] [Green Version]
- Pérez Ortiz, J.M.; Mollema, N.; Toker, N.; Adamski, C.J.; O’Callaghan, B.; Duvick, L.; Friedrich, J.; Walters, M.A.; Strasser, J.; Hawkinson, J.E.; et al. Reduction of protein kinase A-mediated phosphorylation of ATXN1-S776 in Purkinje cells delays onset of Ataxia in a SCA1 mouse model. Neurobiol. Dis. 2018, 116, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Santostefano, K.; Hamazaki, T.; Liu, J.; Subramony, S.H.; Terada, N.; Ashizawa, T. Generation of human-induced pluripotent stem cells to model spinocerebellar ataxia type 2 In Vitro. J. Mol. Neurosci. 2013, 51, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.P.; Sun, X.; Xia, G.; Arbez, N.; Paul, S.; Zhu, S.; Peng, H.B.; Ross, C.A.; Koeppen, A.H.; Margolis, R.L.; et al. ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann. Neurol. 2016, 80, 600–615. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Yang, C.C.; Soong, B.W.; Yu, C.Y.; Chen, S.H.; Huang, H.P.; Kuo, H.C. Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci. Rep. 2019, 9, 1166. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.J.; Golla, M.; Guaitoli, G.; Wanichawan, P.; Hayer, S.N.; Hauser, S.; Krahl, A.C.; Nagel, M.; Samer, S.; Aronica, E.; et al. A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain 2017, 140, 1280–1299. [Google Scholar] [CrossRef]
- Evert, B.O.; Nalavade, R.; Jungverdorben, J.; Matthes, F.; Weber, S.; Rajput, A.; Bonn, S.; Brüstle, O.; Peitz, M.; Krauß, S. Upregulation of miR-370 and miR-543 is associated with reduced expression of heat shock protein 40 in spinocerebellar ataxia type 3. PLoS ONE 2018, 13, e0201794. [Google Scholar] [CrossRef]
- Harmuth, T.; Prell-Schicker, C.; Weber, J.J.; Gellerich, F.; Funke, C.; Drießen, S.; Magg, J.C.D.; Krebiehl, G.; Wolburg, H.; Hayer, S.N.; et al. Mitochondrial Morphology, Function and Homeostasis Are Impaired by Expression of an N-terminal Calpain Cleavage Fragment of Ataxin-3. Front. Mol. Neurosci. 2018, 11, 368. [Google Scholar] [CrossRef]
- Ouyang, S.; Xie, Y.; Xiong, Z.; Yang, Y.; Xian, Y.; Ou, Z.; Song, B.; Chen, Y.; Xie, Y.; Li, H.; et al. CRISPR/Cas9-Targeted Deletion of Polyglutamine in Spinocerebellar Ataxia Type 3-Derived Induced Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 756–770. [Google Scholar] [CrossRef]
- Chen, I.C.; Chang, K.H.; Chen, Y.J.; Chen, Y.C.; Lee-Chen, G.J.; Chen, C.M. Pueraria lobata and Daidzein Reduce Cytotoxicity by Enhancing Ubiquitin-Proteasome System Function in SCA3-iPSC-Derived Neurons. Oxid. Med. Cell Longev. 2019, 2019, 8130481. [Google Scholar] [CrossRef] [Green Version]
- Martier, R.; Sogorb-Gonzalez, M.; Stricker-Shaver, J.; Hübener-Schmid, J.; Keskin, S.; Klima, J.; Toonen, L.J.; Juhas, S.; Juhasova, J.; Ellederova, Z.; et al. Development of an AAV-Based MicroRNA Gene Therapy to Treat Machado-Joseph Disease. Mol. Ther. Methods Clin. Dev. 2019, 15, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, L.S.; Nóbrega, C.; Tavino, S.; Brinkhaus, M.; Matos, C.; Tomé, S.; Moreira, R.; Henriques, D.; Kaspar, B.K.; Pereira de Almeida, L. Ibuprofen enhances synaptic function and neural progenitors proliferation markers and improves neuropathology and motor coordination in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 3691–3703. [Google Scholar] [CrossRef] [PubMed]
- Depla, J.A.; Sogorb-Gonzalez, M.; Mulder, L.A.; Heine, V.M.; Konstantinova, P.; van Deventer, S.J.; Wolthers, K.C.; Pajkrt, D.; Sridhar, A.; Evers, M.M. Cerebral Organoids: A Human Model for AAV Capsid Selection and Therapeutic Transgene Efficacy in the Brain. Mol. Ther. Methods Clin. Dev. 2020, 18, 167–175. [Google Scholar] [CrossRef]
- García-Huerta, P.; Troncoso-Escudero, P.; Wu, D.; Thiruvalluvan, A.; Cisternas-Olmedo, M.; Henríquez, D.R.; Plate, L.; Chana-Cuevas, P.; Saquel, C.; Thielen, P.; et al. Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 2020, 140, 737–764. [Google Scholar] [CrossRef]
- Thiruvalluvan, A.; de Mattos, E.P.; Brunsting, J.F.; Bakels, R.; Serlidaki, D.; Barazzuol, L.; Conforti, P.; Fatima, A.; Koyuncu, S.; Cattaneo, E.; et al. DNAJB6, a Key Factor in Neuronal Sensitivity to Amyloidogenesis. Mol. Cell 2020, 78, 346–358.e349. [Google Scholar] [CrossRef] [PubMed]
- Bavassano, C.A.; Eigentler, R.; Stanika, G.J.; Obermair, S.; Boesch, G.; Dechant, R.; Nat, R. Bicistronic CACNA1A Gene Expression in Neurons Derived from Spinocerebellar Ataxia Type 6 Patient-Induced Pluripotent Stem Cells. Stem Cells Dev. 2017, 26, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Fan, Y.; Zhou, B.; Xu, Z.; Chen, Y.; Sun, X. Generation of induced pluripotent stem cells from skin fibroblasts of a patient with olivopontocerebellar atrophy. Tohoku J. Exp. Med. 2012, 226, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.M.; Stoyas, C.A.; Switonski, P.M.; Ichou, F.; Fan, W.; Collins, B.; Wall, C.E.; Adanyeguh, I.; Niu, C.; Sopher, B.L.; et al. Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cell Rep. 2019, 26, 1189–1202.e6. [Google Scholar] [CrossRef] [Green Version]
- Burman, R.J.; Watson, L.M.; Smith, D.C.; Raimondo, J.V.; Ballo, R.; Scholefield, J.; Cowley, S.A.; Wood, M.J.A.; Kidson, S.H.; Greenberg, L.J. Molecular and electrophysiological features of spinocerebellar ataxia type seven in induced pluripotent stem cells. PLoS ONE 2021, 16, e0247434. [Google Scholar] [CrossRef]
- Wong, M.M.K.; Hoekstra, S.D.; Vowles, J.; Watson, L.M.; Fuller, G.; Németh, A.H.; Cowley, S.A.; Ansorge, O.; Talbot, K.; Becker, E.B.E. Neurodegeneration in SCA14 is associated with increased PKCγ kinase activity, mislocalization and aggregation. Acta Neuropathol Commun. 2018, 6, 99. [Google Scholar] [CrossRef]
- Schuster, S.; Heuten, E.; Velic, A.; Admard, J.; Synofzik, M.; Ossowski, S.; Macek, B.; Hauser, S.; Schöls, L. CHIP mutations affect the heat shock response differently in human fibroblasts and iPSC-derived neurons. Dis. Models Mech. 2020, 13, dmm045096. [Google Scholar] [CrossRef]
- Matsuzono, K.; Imamura, K.; Murakami, N.; Tsukita, K.; Yamamoto, T.; Izumi, Y.; Kaji, R.; Ohta, Y.; Yamashita, T.; Abe, K.; et al. Antisense Oligonucleotides Reduce RNA Foci in Spinocerebellar Ataxia 36 Patient iPSCs. Mol. Ther. Nucleic Acids 2017, 8, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hommersom, M.P.; Buijsen, R.A.M.; van Roon-Mom, W.M.C.; van de Warrenburg, B.P.C.; van Bokhoven, H. Human Induced Pluripotent Stem Cell-Based Modelling of Spinocerebellar Ataxias. Stem Cell Rev. Rep. 2021. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Shuvalova, L.D.; Davidenko, A.V.; Eremeev, A.V.; Khomyakova, E.A.; Zerkalenkova, E.A.; Lebedeva, O.S.; Bogomazova, A.N.; Klyushnikov, S.A.; Illarioshkin, S.N.; Lagarkova, M.A. Generation of induced pluripotent stem cell line RCPCMi008-A derived from patient with spinocerebellar ataxia 17. Stem Cell Res. 2021, 54, 102431. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [Green Version]
- Boulting, G.L.; Kiskinis, E.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; Wainger, B.J.; Williams, D.J.; Kahler, D.J.; Yamaki, M.; Davidow, L.; et al. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 279–286. [Google Scholar] [CrossRef]
- Mitne-Neto, M.; Machado-Costa, M.; Marchetto, M.C.; Bengtson, M.H.; Joazeiro, C.A.; Tsuda, H.; Bellen, H.J.; Silva, H.C.; Oliveira, A.S.; Lazar, M.; et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 2011, 20, 3642–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Goparaju, S.K.; Tooi, N.; Inoue, H.; Takahashi, R.; Nakatsuji, N.; Aiba, K. Amyotrophic lateral sclerosis model derived from human embryonic stem cells overexpressing mutant superoxide dismutase 1. Stem. Cells Transl. Med. 2012, 1, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A Cellular Model for Sporadic ALS Using Patient-Derived Induced Pluripotent Stem Cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W., 4th; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.; Li, D.; Louis, K.R.; Li, X.; Yang, H.; Sun, Q.; Crandall, S.R.; Tsang, S.; Zhou, J.; Cox, C.L.; et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 2014, 5, 3449. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.L.; Zhong, X.; Fan, F.; Zhang, S.C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [Green Version]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Inoue, H. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [Green Version]
- Bursch, F.; Kalmbach, N.; Naujock, M.; Staege, S.; Eggenschwiler, R.; Abo-Rady, M.; Japtok, J.; Guo, W.; Hensel, N.; Reinhardt, P.; et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 2019, 28, 2835–2850. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell. 2017, 8, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiyanagi, N.; Fujimori, K.; Yano, M.; Ishihara-Fujisaki, C.; Sone, T.; Akiyama, T.; Okada, Y.; Akamatsu, W.; Matsumoto, T.; Ishikawa, M.; et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 496–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [Green Version]
- Tyzack, G.E.; Hall, C.E.; Sibley, C.R.; Cymes, T.; Forostyak, S.; Carlino, G.; Meyer, I.F.; Schiavo, G.; Zhang, S.C.; Gibbons, G.M.; et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat. Commun. 2017, 8, 1164. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Li, Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: A systematic review. Mol. Brain 2020, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Bekdash, R.; Klein, A.D.; Yazawa, M. Timothy syndrome iPSC modeling. Mol. Cell Neurosci. 2020, 107, 103529. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Paşca, S.P.; Shcheglovitov, A.; Yazawa, M.; Schwemberger, R.; Rasmusson, R.; Dolmetsch, R.E. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 2013, 16, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Paşca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Paşca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Panagiotakos, G.; Haveles, C.; Arjun, A.; Petrova, R.; Rana, A.; Portmann, T.; Paşca, S.P.; Palmer, T.D.; Dolmetsch, R.E. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife 2019, 8, e51037. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Díaz, F.; Canals, I.; Hansen, M.G.; Rufián-Vázquez, L.; Ehinger, J.K.; Elmér, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. Int. J. Mol. Sci. 2020, 21, 3191. [Google Scholar] [CrossRef] [PubMed]
- Avazzadeh, S.; McDonagh, K.; Reilly, J.; Wang, Y.; Boomkamp, S.D.; McInerney, V.; Krawczyk, J.; Fitzgerald, J.; Feerick, N.; O’Sullivan, M.; et al. Increased Ca2+ signaling in NRXN1α +/- neurons derived from ASD induced pluripotent stem cells. Mol. Autism 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
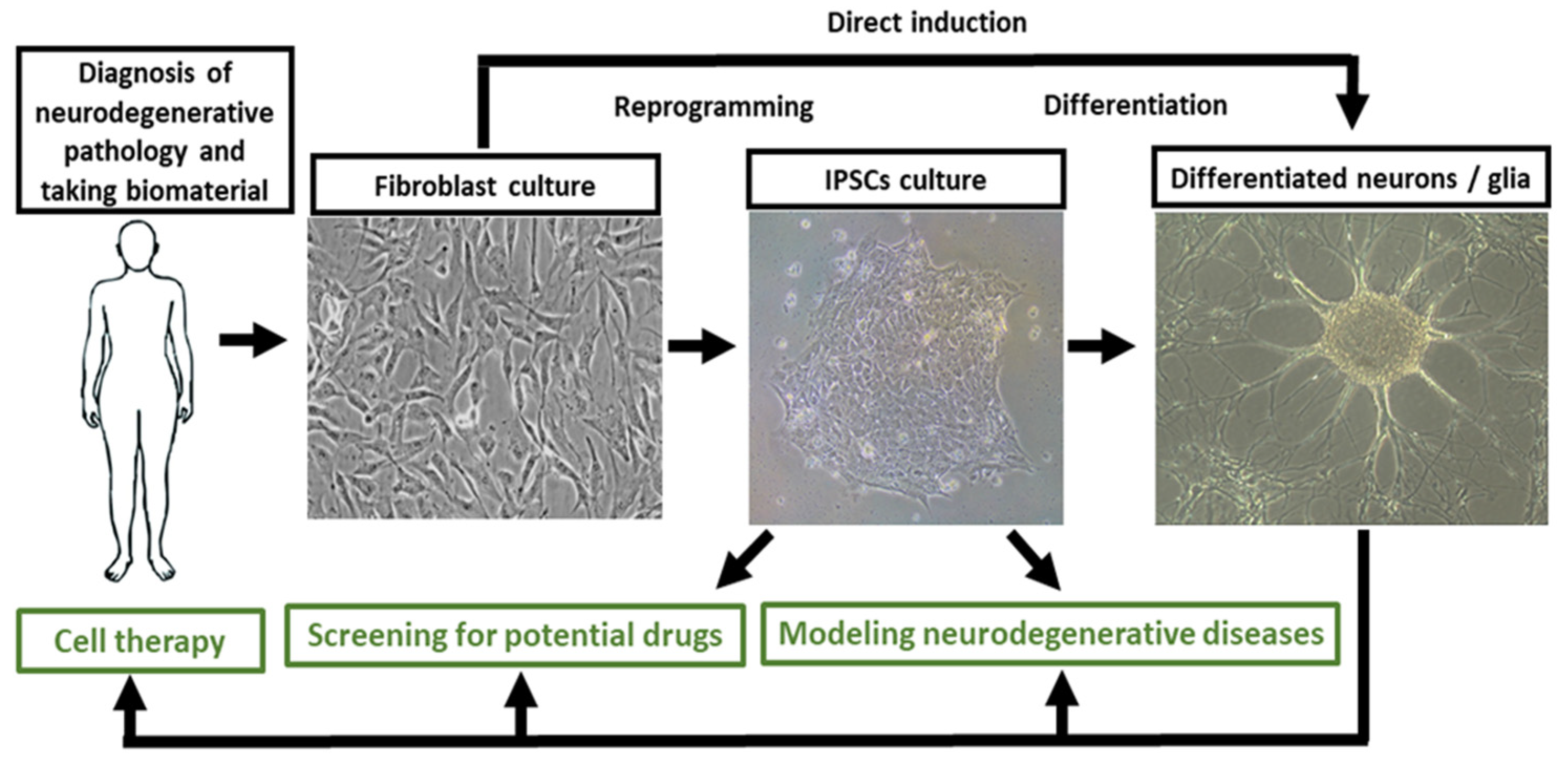

{kind=link}
| Alzheimer’s Disease | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
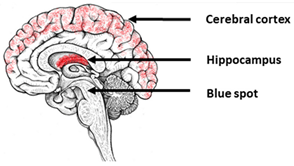 | Patient-specific models | ||
| Neurons | [37,46,48,97,99,100,101,102,103,104] | ||
| Cortical neurons | [105,106] | ||
| Cholinergic neurons | [107] | ||
| Astrocytes | [105,108] | ||
| Endothelial cells | [72,109] | ||
| Microglia | [67,69] | ||
| Isogenic models | |||
| Neurons | [37,38,39,98] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| PSEN1 | Neurons | Increased ROS production, impaired energy status and mitochondrial potential, impaired autophagy, ER stress and increased calcium release from the ER | [104] |
| Astrocytes | Disturbed calcium release from the ER, increased ROS production | [108] | |
| PSEN2 | Neurons | Increased amplitude of spontaneous calcium oscillations and their desynchronization, hyperactivation of neurons | [99] |
| APP | Astrocytes | ER stress, increased ROS production | [105] |
| Sporadic forms | Neurons | Mitochondrial dysfunction, increased ROS production, increased levels of oxidative phosphorylation chain complexes | [103] |
| Parkinson’s Disease | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
 | Patient-specific models | ||
| NESC | [117] | ||
| Dopaminergic neurons (DAn) | [51,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,138] | ||
| Cortical neurons | [137] | ||
| Astrocytes | [66] | ||
| Microglia | [68] | ||
| Isogenic models | |||
| DAn | [41,45] | ||
| Cortical neurons | [43] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| LRRK2 | NESC | Mitochondrial dysfunction, impaired mitophagy | [117] |
| DAn | Enhanced expression of genes involved in calcium signaling, as well as an increase in calcium influx, mitochondrial dysfunction, mitochondrial DNA damage, oxidative stress, disrupted calcium dynamics in the ER | [120,121,124,125,127,131,132,136,138] | |
| SNCA | Cortical neurons | Mitochondrial dysfunction, oxidative stress | [137] |
| DAn | Mitochondrial dysfunction, ER stress, impaired calcium homeostasis and ATP production | [43,119,130,131,135,139] | |
| PINK1 | DAn | Mitochondrial dysfunction, oxidative stress, slowed utilization of damaged mitochondria | [133] |
| PARK2 | DAn | Mitochondrial dysfunction, oxidative stress, dysfunction of voltage-gated T-type calcium channels | [41,51,134,140] |
| DJ-1 | DAn | Oxidative stress | [129] |
| Huntington’s Disease | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
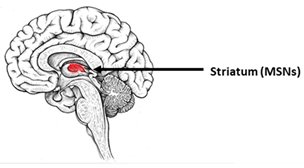 | Patient-specific models | ||
| iPSCs | [50] | ||
| NPC | [52,152,153,161] | ||
| Cortical neurons | [154] | ||
| GABAergic neurons | [8,61,155,156,157,158] | ||
| Astrocytes | [159,160] | ||
| Endothelial cells | [71] | ||
| Mixed cultures | [73] | ||
| Isogenic models | |||
| NPC | [44] | ||
| GABAergic neurons | [36] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| HTT | iPSCs | Impaired expression of the genes responsible for cell death | [50] |
| Mixed cultures | Impaired expression of genes responsible for calcium signaling | [73] | |
| GABAergic neurons | Increased store-operated calcium entry and calcium currents through voltage-gated calcium channels in low-repet and juvenile models of Huntington’s disease | [8,61,157] | |
| GABAergic neurons | Increased mitochondrial density | [156] | |
| GABAergic neurons | Dysfunction of lysosomes and autophagy | [8,155] | |
| NPC | Mitochondrial dysfunction, bioenergetic defects | [44,161] | |
| Spinocerebellar Ataxias | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
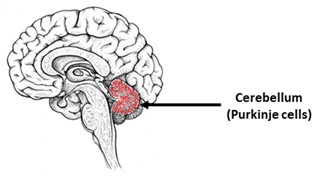 | Patient-specific models | ||
| iPSCs | [193] | ||
| NPC | [176,177] | ||
| Neurons | [58,59,60,174,175,180,182,183,184,185,186,187,188,190,192] | ||
| Cortical neurons | [179,181,194] | ||
| Motoneurons (MNs) | [195] | ||
| GABAergic and glutamatergic neurons | [189] | ||
| Purkinje cells | [167,170,171,172,173] | ||
| Mixed cultures | [178] | ||
| Isogenic models | |||
| NPC | [191] | ||
| Purkinje cells | [42] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| ATXN1(SCA1) | GABAergic neurons | Decreased store-operated calcium entry, no changes in the functioning of voltage-gated calcium channels | Vigont et al., unpublished data |
| ATXN2 & 3(SCA2 & 3) | Mixed cultures | Altered expression of glutamate receptor subunits and other participants in calcium signaling. Increased calcium concentration in the cytosol. Distorted mitochondrial microstructures and mitochondrial dysfunction | [178] |
| ATXN3(SCA3) | Cortical neurons | Mitochondrial fragmentation and cristae alterations leading to decreased capacity of mitochondrial respiration, impaired mitochondrial degradation. Enhanced calpains-activated ataxin 3 cleavage and produce ataxin 3 fragments. | [58,179,181] |
| CACNA1A (SCA6) | GABAergic& glutamatergic neurons | No differences in both CACNA1A expression and amplitudes of voltage-gated calcium currents | [189] |
| ATXN7(SCA7) | NPC | Decreased mitochondrial network length. Mitochondrial dysfunction, reduced oxygen consumption rate and increased extracellular acidification rate | [191] |
| TBP(SCA17) | GABAergic neurons | Increased store-operated calcium entry and voltage-gated calcium uptake | Vigont et al., unpublished data |
| Amyotrophic Lateral Sclerosis | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
 | Patient-specific models | ||
| Motoneurons (MNs) | [64,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219] | ||
| Cortical neurons | [205] | ||
| Astrocytes | [202,224,225] | ||
| Oligodendrocyte | [226] | ||
| Isogenic models | |||
| Motoneurons (MNs) | [43] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| C9orf72 | MNs | Transcriptional changes in levels of the mitochondrial transporter, increased expression of genes encoding glutamate receptors and VGCC. Increased glutamate excitotoxicity C9ORF72 iPSN cultures. Disrupted mitochondrial calcium buffering capacity, low calbindin levels | [204,217,218,219,221] |
| TARDBP | MNs | Increased expression of genes encoding glutamate receptors. Elevated basal intracellular calcium levels. Increased conductance for calcium AMPA and NMDA receptors. Imbalance in mitochondrial calcium buffering capacity | [217,218,219] |
| SOD1 | MNs | Transcriptional and functional changes: defects in mitochondrial transport, morphology and motility, increase in mitochondrial density, oxidative and ER-related stress. Reduced level of calcium-binding proteins. | [220,221] |
| FUS | MNs | No morphological changes in the mitochondria, decreased mitochondrial motility | [223] |
| Sporadic form | MNs | Deregulated expression of the genes associated with mitochondrial functioning | [209] |
| Other Neurological Pathologies | |||
|---|---|---|---|
| The most vulnerable areas of the brain | iPSCs-based models | ||
 | Patient-specific models | ||
| NPC (LS) | [237] | ||
| Neurons (ASD) | [234,235,236] | ||
| Cortical neurons (ASD) | [238] | ||
| Mutant genes | Cell type | Disturbance associated with calcium signaling | |
| Genes encoding VGCC subunits (ASD) | Neurons | Altered expression of genes responsible for calcium signaling. Dysfunction of voltage-gated channels. Increased nimodipine sensitive calcium influx after depolarization. | [234,235] |
| mtDNA m.13513G (LS) | NPC | Mitochondrial dysfunction, decreased calcium buffering capacity | [237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grekhnev, D.A.; Kaznacheyeva, E.V.; Vigont, V.A. Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling. Int. J. Mol. Sci. 2022, 23, 624. https://doi.org/10.3390/ijms23020624
Grekhnev DA, Kaznacheyeva EV, Vigont VA. Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling. International Journal of Molecular Sciences. 2022; 23(2):624. https://doi.org/10.3390/ijms23020624
Chicago/Turabian StyleGrekhnev, Dmitriy A., Elena V. Kaznacheyeva, and Vladimir A. Vigont. 2022. "Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling" International Journal of Molecular Sciences 23, no. 2: 624. https://doi.org/10.3390/ijms23020624
APA StyleGrekhnev, D. A., Kaznacheyeva, E. V., & Vigont, V. A. (2022). Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling. International Journal of Molecular Sciences, 23(2), 624. https://doi.org/10.3390/ijms23020624