
Silibinin Suppresses the Hyperlipidemic Effects of the ALK-Tyrosine Kinase Inhibitor Lorlatinib in Hepatic Cells

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
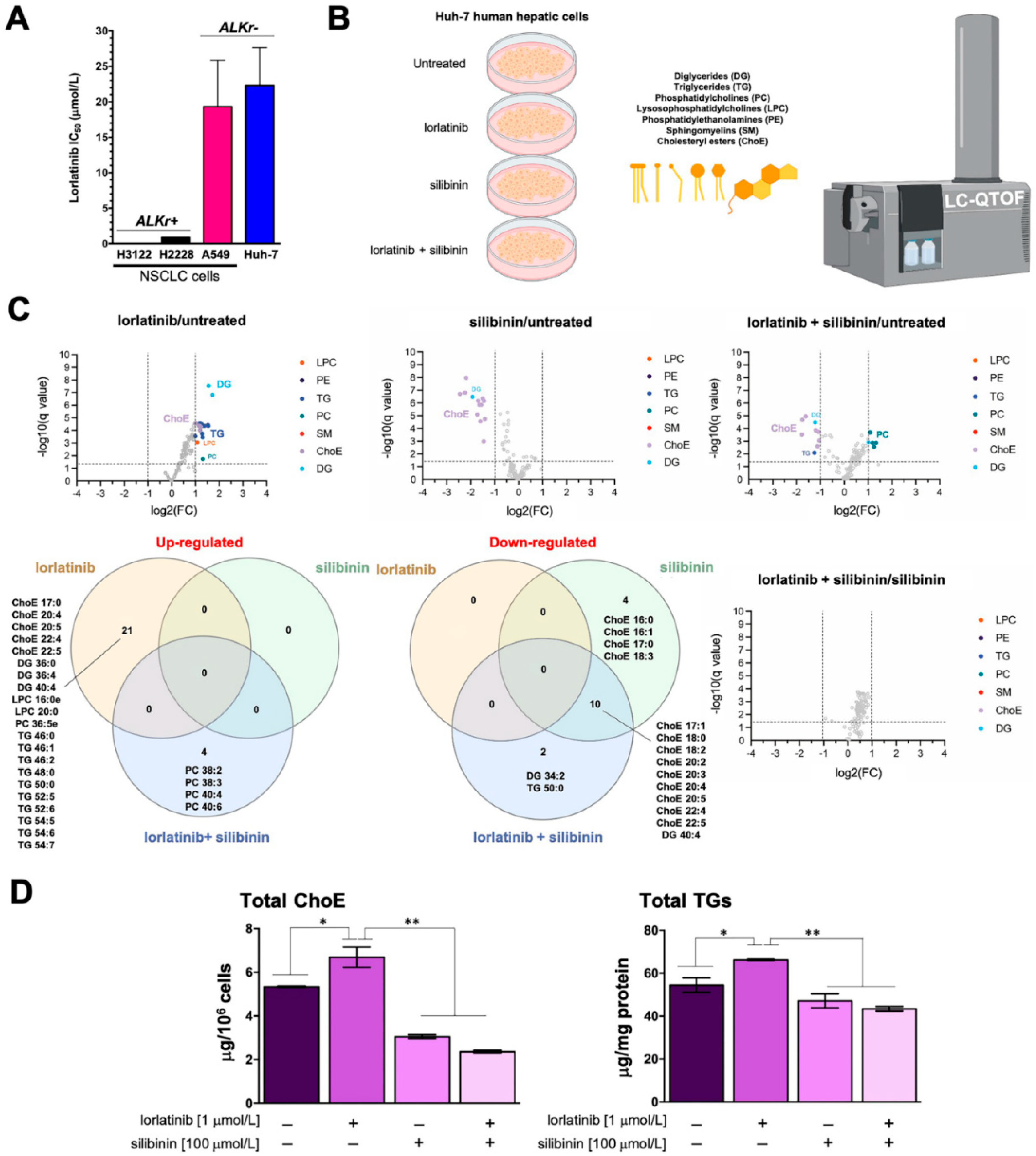
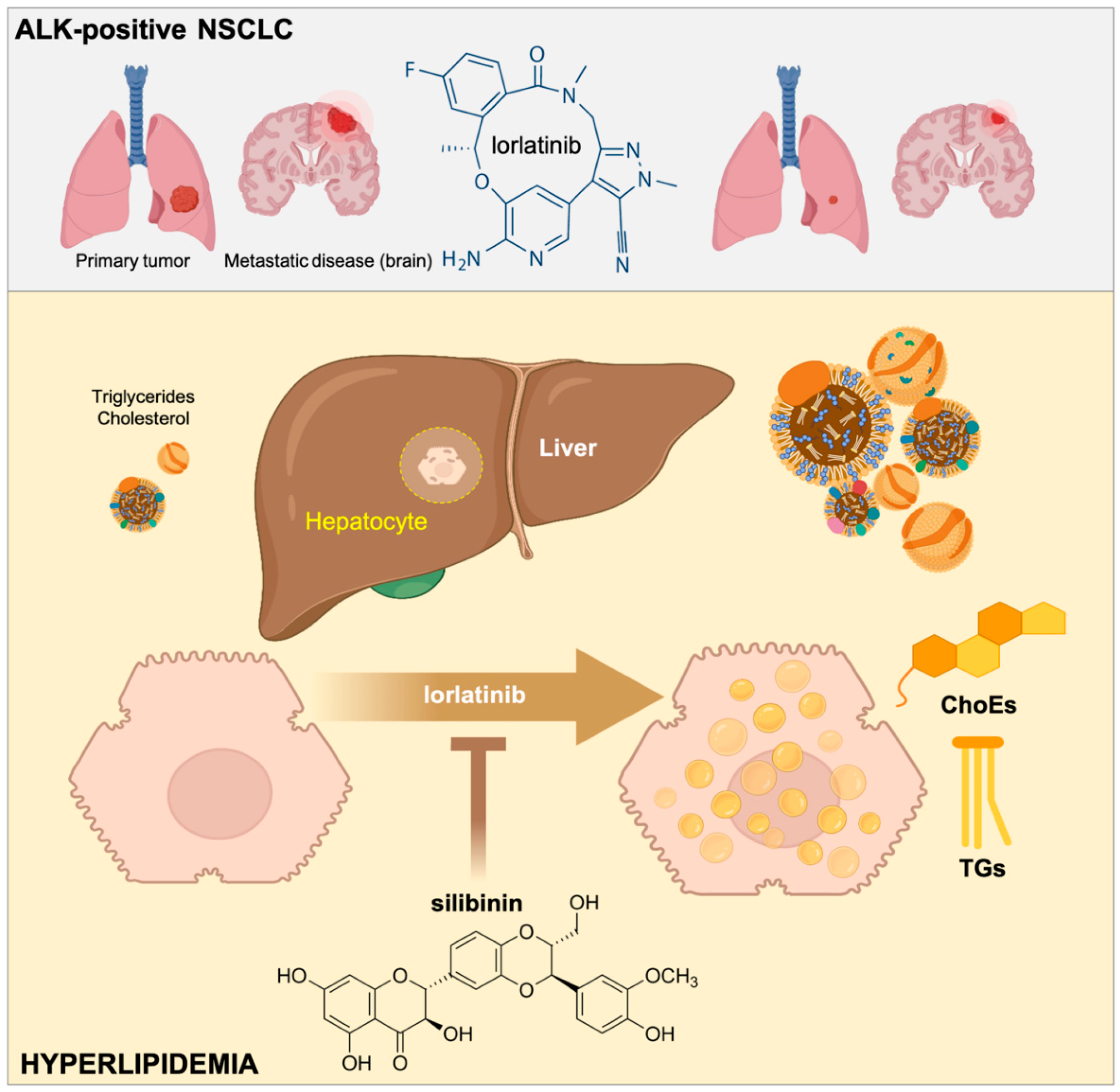
2.1. Hepatic Cells Treated with Lorlatinib Accumulate Cholesteryl Esters and Triglycerides
2.2. Silibinin Fully Protects the Steady-State Lipidome of Hepatic Cells against the Hyperlipidemic Effects of Lorlatinib
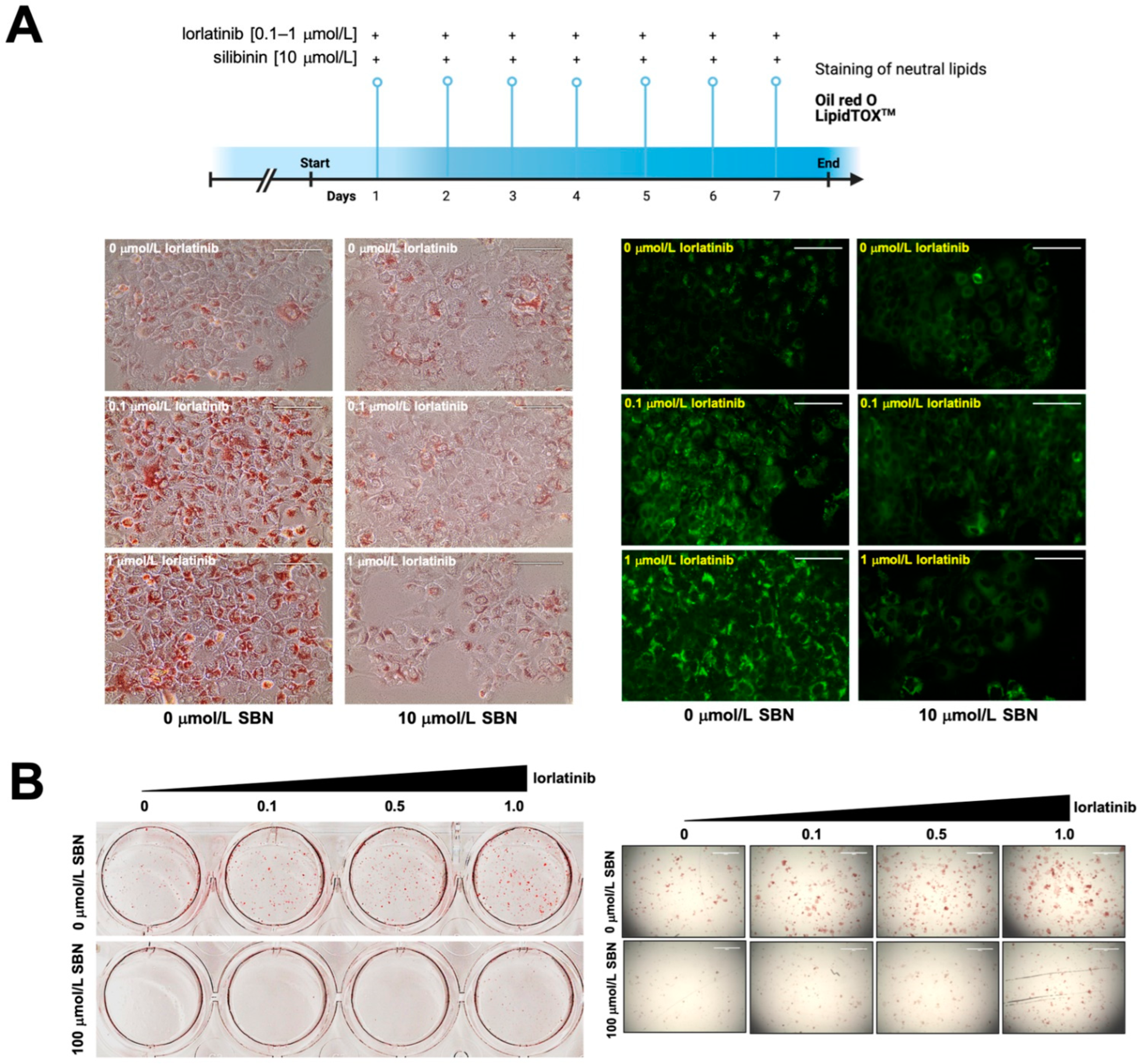
2.3. Silibinin Prevents the Lorlatinib-Induced Chronic Accumulation of Neutral Lipids in Hepatic Cell
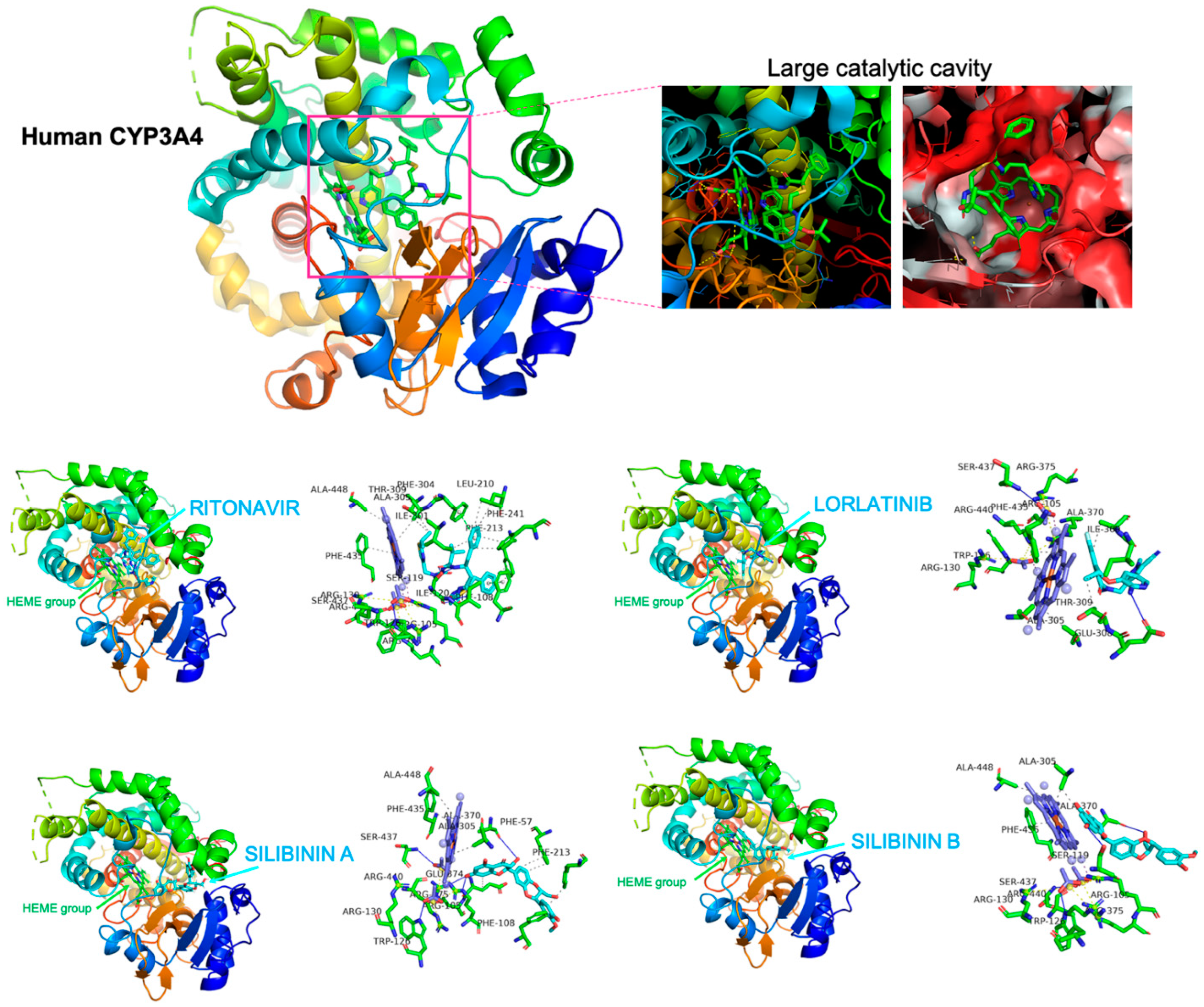
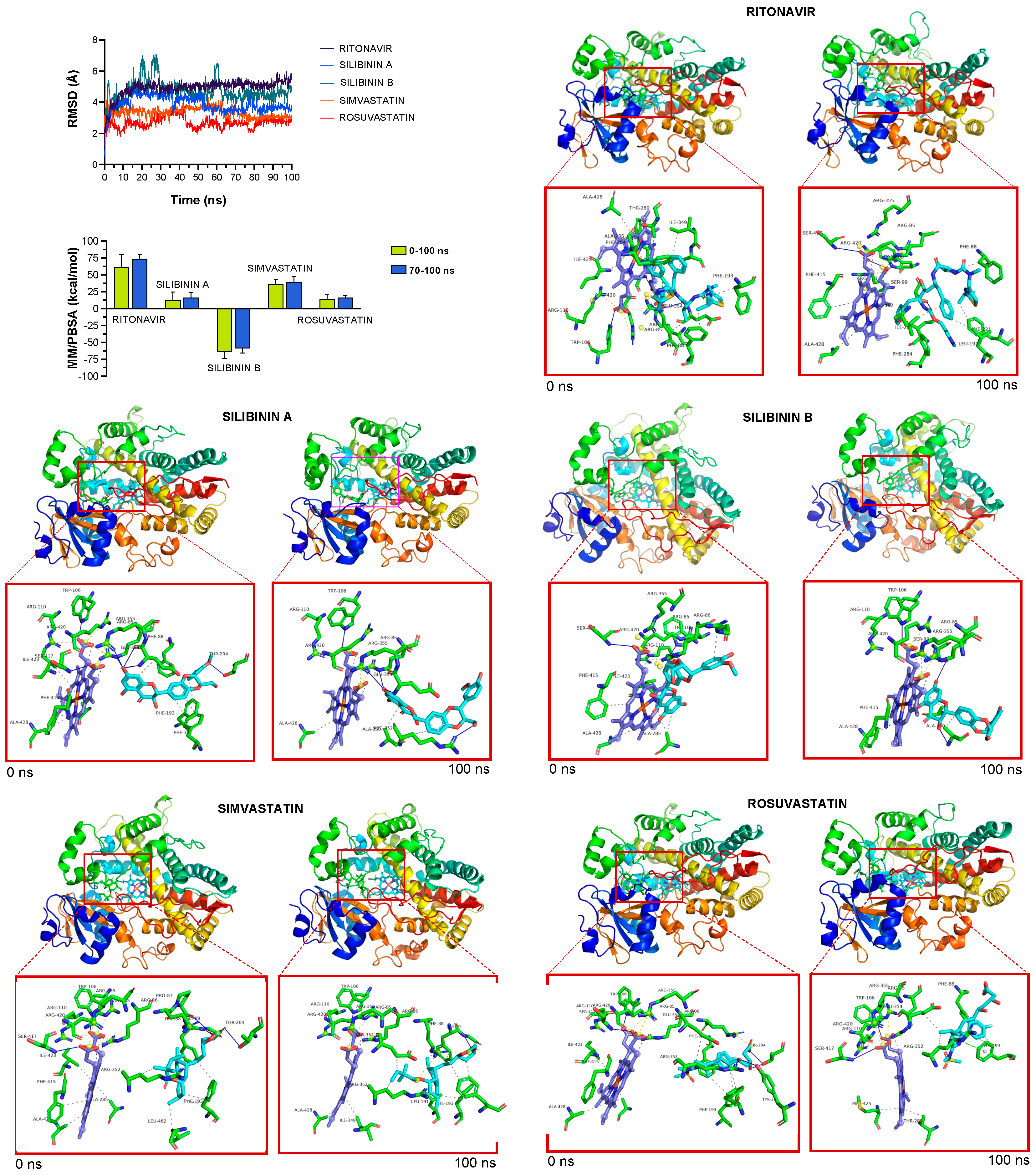
2.4. Silibinin Does Not Overlap the Binding Mode of Lorlatinib to Cytochrome P450 3A4 (CYP3A4) In Silico
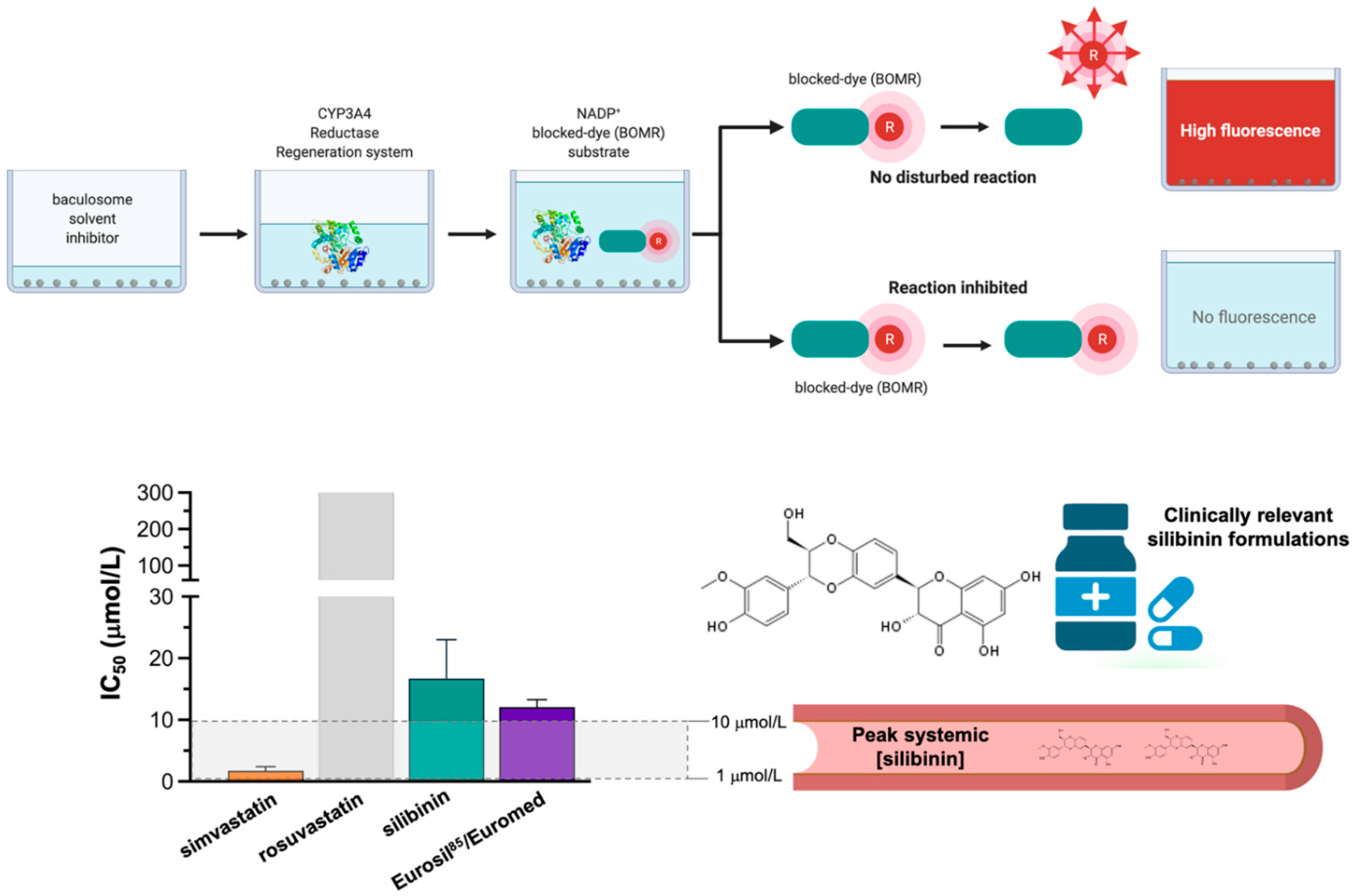
2.5. Silibinin Is a Weak Inhibitor of the Lorlatinib-Metabolizing Cytochrome P450 3A4 (CYP3A4) Isoenzyme
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines
4.3. Cell Viability Assays
4.4. Non-Targeted Lipidomics
4.5. Triglyceride and Cholesterol Quantification
4.6. Accumulation of Neutral Lipids
4.7. Prediction of CYP450 Inhibition In Silico
4.8. Docking Calculations, Molecular Dynamics Simulations, and Binding Free Energy Analysis
4.9. CYP3A4 Inhibition
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhu, L.; Sun, Y.; Stebbing, J.; Selvaggi, G.; Zhang, Y.; Yu, Z. Targeting ALK Rearrangements in NSCLC: Current State of the Art. Front. Oncol. 2022, 12, 863461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, W. Lorlatinib for the treatment of anaplastic lymphoma kinase-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 173–178. [Google Scholar] [CrossRef]
- Choo, J.R.; Soo, R.A. Lorlatinib for the treatment of ALK-positive metastatic non-small cell lung cancer. Expert Rev. Anticancer Ther. 2020, 20, 233–240. [Google Scholar] [CrossRef]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.L.; Dinh, D.; et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo [4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar]
- Collier, T.L.; Normandin, M.D.; Stephenson, N.A.; Livni, E.; Liang, S.H.; Wooten, D.W.; Esfahani, S.A.; Stabin, M.G.; Mahmood, U.; Chen, J.; et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat. Commun. 2017, 8, 15761. [Google Scholar] [CrossRef]
- Costa, D.B.; Shaw, A.T.; Ou, S.H.; Solomon, B.J.; Riely, G.J.; Ahn, M.J.; Zhou, C.; Shreeve, S.M.; Selaru, P.; Polli, A.; et al. Clinical Experience with Crizotinib in Patients with Advanced ALK-Rearranged Non-Small-Cell Lung Cancer and Brain Metastases. J. Clin. Oncol. 2015, 33, 1881–1888. [Google Scholar] [CrossRef]
- Bauer, T.M.; Shaw, A.T.; Johnson, M.L.; Navarro, A.; Gainor, J.F.; Thurm, H.; Pithavala, Y.K.; Abbattista, A.; Peltz, G.; Felip, E. Brain Penetration of Lorlatinib: Cumulative Incidences of CNS and Non-CNS Progression with Lorlatinib in Patients with Previously Treated ALK-Positive Non-Small-Cell Lung Cancer. Target Oncol. 2020, 15, 55–65. [Google Scholar] [CrossRef]
- Felip, E.; Shaw, A.T.; Bearz, A.; Camidge, D.R.; Solomon, B.J.; Bauman, J.R.; Bauer, T.M.; Peters, S.; Toffalorio, F.; Abbattista, A.; et al. Intracranial and extracranial efficacy of lorlatinib in patients with ALK-positive non-small-cell lung cancer previously treated with second-generation ALK TKIs. Ann. Oncol. 2021, 32, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.M.; Bazhenova, L.A. Update on Lorlatinib: Role in Reducing the Risk of Disease Progression in ALK-Positive NSCLC. Cancer Manag. Res. 2022, 14, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Lorlatinib: First Global Approval. Drugs 2019, 79, 93–98. [Google Scholar] [CrossRef]
- Bauer, T.M.; Felip, E.; Solomon, B.J.; Thurm, H.; Peltz, G.; Chioda, M.D.; Shaw, A.T. Clinical Management of Adverse Events Associated with Lorlatinib. Oncologist 2019, 24, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Blais, N.; Adam, J.-P.; Nguyen, J.; Grégoire, J.C. Evaluation and Management of Dyslipidemia in Patients Treated with Lorlatinib. Curr. Oncol. 2021, 28, 265–272. [Google Scholar] [CrossRef]
- Neuvonen, P.J.; Niemi, M.; Backman, J.T. Drug interactions with lipid-lowering drugs: Mechanisms and clinical relevance. Clin. Pharmacol. Ther. 2006, 80, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Rosales, A.S.; Chioda, M.D.; Parker, L.; Devgan, G.; Kettle, J. Consensus Recommendations for Management and Counseling of Adverse Events Associated With Lorlatinib: A Guide for Healthcare Practitioners. Adv. Ther. 2020, 37, 3019–3030. [Google Scholar] [CrossRef]
- Fernández-Arroyo, S.; Hernández-Aguilera, A.; de Vries, M.A.; Burggraaf, B.; van der Zwan, E.; Pouw, N.; Joven, J.; Cabezas, M.C. Effect of Vitamin D3 on the Postprandial Lipid Profile in Obese Patients: A Non-Targeted Lipidomics Study. Nutrients 2019, 11, 1194. [Google Scholar] [CrossRef]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar]
- Kawamoto, M.; Yamaji, T.; Saito, K.; Shirasago, Y.; Satomura, K.; Endo, T.; Fukasawa, M.; Hanada, K.; Osada, N. Identification of Characteristic Genomic Markers in Human Hepatoma HuH-7 and Huh7.5.1-8 Cell Lines. Front. Genet. 2020, 11, 546106. [Google Scholar] [CrossRef]
- Ramboer, E.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Immortalized Human Hepatic Cell Lines for In Vitro Testing and Research Purposes. Methods Mol. Biol. 2015, 1250, 53–76. [Google Scholar] [PubMed]
- Berger, E.; Vega, N.; Weiss-Gayet, M.; Géloën, A. Gene Network Analysis of Glucose Linked Signaling Pathways and Their Role in Human Hepatocellular Carcinoma Cell Growth and Survival in HuH7 and HepG2 Cell Lines. Biomed. Res. Int. 2015, 2015, 821761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulutoglu, B.; Rey-Bedón, C.; Mert, S.; Tian, L.; Jang, Y.Y.; Yarmush, M.L.; Usta, O.B. A comparison of hepato-cellular in vitro platforms to study CYP3A4 induction. PLoS ONE 2020, 15, e0229106. [Google Scholar] [CrossRef] [PubMed]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M. Silybin, a Major Bioactive Component of Milk Thistle (Silybum marianum L. Gaernt.)-Chemistry, Bioavailability, and Metabolism. Molecules 2017, 22, 1942. [Google Scholar] [CrossRef]
- Křen, V.; Valentová, K. Silybin and its congeners: From traditional medicine to molecular effects. Nat. Prod. Rep. 2022, 39, 1264–1281. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Wu, S.C. Health Benefits of Silybum marianum: Phytochemistry, Pharmacology, and Applications. J. Agric. Food Chem. 2020, 68, 11644–11664. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef]
- Hackett, E.S.; Twedt, D.C.; Gustafson, D.L. Milk thistle and its derivative compounds: A review of opportunities for treatment of liver disease. J. Vet. Intern. Med. 2013, 27, 10–16. [Google Scholar] [CrossRef]
- Abenavoli, L.; Capasso, R.; Milic, N.; Capasso, F. Milk thistle in liver diseases: Past, present, future. Phytother. Res. 2010, 24, 1423–1432. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Liu, J.; Zha, J.; Pei, L. Evaluation of EML4-ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia 2011, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, E.R.; Sevrioukova, I.F. Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength. Int. J. Mol. Sci. 2021, 22, 852. [Google Scholar] [CrossRef] [PubMed]
- Domanski, T.L.; He, Y.A.; Harlow, G.R.; Halpert, J.R. Dual role of human cytochrome P450 3A4 residue Phe-304 in substrate specificity and cooperativity. J. Pharmacol. Exp. Ther. 2000, 293, 585–591. [Google Scholar]
- Hackett, J.C. Membrane-embedded substrate recognition by cytochrome P450 3A4. J. Biol. Chem. 2018, 293, 4037–4046. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr. Top. Med. Chem. 2014, 14, 1348–1355. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef]
- Gillessen, A.; Schmidt, H.H. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv. Ther. 2020, 37, 1279–1301. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef] [PubMed]
- Schnell, P.; Bartlett, C.H.; Solomon, B.J.; Tassell, V.; Shaw, A.T.; de Pas, T.; Lee, S.H.; Lee, G.K.; Tanaka, K.; Tan, W.; et al. Complex renal cysts associated with crizotinib treatment. Cancer Med. 2015, 4, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, E.E.; Usari, T.; Polli, A.; Lewis, I.; Wilner, K.D. Renal effects of crizotinib in patients with ALK-positive advanced NSCLC. J. Thorac. Oncol. 2019, 14, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.H. Nephrotoxicity of cancer therapeutic drugs: Focusing on novel agents. Kidney Res. Clin. Pract. 2021, 40, 344–354. [Google Scholar] [CrossRef] [PubMed]
- McGee, K.; Stone, N.J.; Wadhwani, S.; Kanwar, Y.S.; Villaflor, V.; Akhter, N. A possible mechanism of hyperlipidemia in a patient with metastatic non-small cell lung cancer on lorlatinib therapy. J. Oncol. Pharm. Pract. 2021, 27, 2010–2013. [Google Scholar] [CrossRef]
- Hibma, J.E.; O’Gorman, M.; Nepal, S.; Pawlak, S.; Ginman, K.; Pithavala, Y.K. Evaluation of the absolute oral bioavailability of the anaplastic lymphoma kinase/c-ROS oncogene 1 kinase inhibitor lorlatinib in healthy participants. Cancer Chemother. Pharmacol. 2022, 89, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Houk, B.; Pithavala, Y.K.; Ruiz-Garcia, A. Population pharmacokinetic model with time-varying clearance for lorlatinib using pooled data from patients with non-small cell lung cancer and healthy participants. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 148–160. [Google Scholar] [CrossRef]
- Chen, W.; Shi, Y.; Qi, S.; Zhou, H.; Li, C.; Jin, D.; Li, G. Pharmacokinetic Study and Tissue Distribution of Lorlatinib in Mouse Serum and Tissue Samples by Liquid Chromatography-Mass Spectrometry. J. Anal. Methods Chem. 2019, 2019, 7574369. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, M.A.; de la Cruz-Ojeda, P.; López-Grueso, M.J.; Navarro-Villarán, E.; Requejo-Aguilar, R.; Castejón-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, Á.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Vinken, M. Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. Int. J. Mol. Sci. 2022, 23, 3315. [Google Scholar] [CrossRef]
- Yin, F.; Gupta, R.; Vergnes, L.; Driscoll, W.S.; Ricks, J.; Ramanathan, G.; Stewart, J.A.; Shih, D.M.; Faull, K.F.; Beaven, S.W.; et al. Diesel Exhaust Induces Mitochondrial Dysfunction, Hyperlipidemia, and Liver Steatosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1776–1786. [Google Scholar] [CrossRef]
- Remon, J.; Besse, B. Brain Metastases in Oncogene-Addicted Non-Small Cell Lung Cancer Patients: Incidence and Treatment. Front Oncol. 2018, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Sais, E.; Cañete, N.; Marruecos, J.; Cuyàs, E.; Izquierdo, A.; Porta, R.; Haro, M.; Brunet, J.; Pedraza, S.; et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget 2016, 7, 32006–32014. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón, Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) restricts brain accumulation and cytochrome P450-3A (CYP3A) limits oral availability of the novel ALK/ROS1 inhibitor lorlatinib. Int. J. Cancer 2018, 143, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lettiere, D.; Tse, S.; Johnson, T.R.; Biddle, K.E.; Thibault, S.; Palazzi, X.; Chen, J.; Pithavala, Y.K.; Finkelstein, M. Liver Toxicity Observed With Lorlatinib When Combined With Strong CYP3A Inducers: Evaluation of Cynomolgus Monkey as a Nonclinical Model for Assessing the Mechanism of Combinational Toxicity. Toxicol. Sci. 2021, 182, 183–194. [Google Scholar] [CrossRef]
- Brantley, S.J.; Graf, T.N.; Oberlies, N.H.; Paine, M.F. A systematic approach to evaluate herb-drug interaction mechanisms: Investigation of milk thistle extracts and eight isolated constituents as CYP3A inhibitors. Drug Metab. Dispos. 2013, 41, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Beckmann-Knopp, S.; Rietbrock, S.; Weyhenmeyer, R.; Böcker, R.H.; Beckurts, K.T.; Lang, W.; Hunz, M.; Fuhr, U. Inhibitory effects of silibinin on cytochrome P-450 enzymes in human liver microsomes. Pharmacol. Toxicol. 2000, 86, 250–256. [Google Scholar] [CrossRef]
- Lin, J.; Schyschka, L.; Mühl-Benninghaus, R.; Neumann, J.; Hao, L.; Nussler, N.; Dooley, S.; Liu, L.; Stöckle, U.; Nussler, A.K.; et al. Comparative analysis of phase I and II enzyme activities in 5 hepatic cell lines identifies Huh-7 and HCC-T cells with the highest potential to study drug metabolism. Arch. Toxicol. 2012, 86, 87–95. [Google Scholar] [CrossRef]
- Liu, Y.; Flynn, T.J.; Xia, M.; Wiesenfeld, P.L.; Ferguson, M.S. Evaluation of CYP3A4 inhibition and hepatotoxicity using DMSO-treated human hepatoma HuH-7 cells. Cell. Biol. Toxicol. 2015, 31, 221–230. [Google Scholar] [CrossRef]
- Guo, L.; Dial, S.; Branham, W.; Liu, J.; Fang, J.L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Sivertsson, L.; Ek, M.; Darnell, M.; Edebert, I.; Ingelman-Sundberg, M.; Neve, E.P. CYP3A4 catalytic activity is induced in confluent Huh7 hepatoma cells. Drug Metab. Dispos. 2010, 38, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Sivertsson, L.; Edebert, I.; Palmertz, M.P.; Ingelman-Sundberg, M.; Neve, E.P. Induced CYP3A4 expression in confluent Huh7 hepatoma cells as a result of decreased cell proliferation and subsequent pregnane X receptor activation. Mol. Pharmacol. 2013, 83, 659–670. [Google Scholar] [CrossRef]
- Kanebratt, K.P.; Andersson, T.B. HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG cell line: A unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Cuyàs, E.; Lozano-Sánchez, J.; Cufí, S.; Verdura, S.; Fernández-Arroyo, S.; Borrás-Linares, I.; Martin-Castillo, B.; Martin, Á.G.; Lupu, R.; et al. Extra-virgin olive oil contains a metabolo-epigenetic inhibitor of cancer stem cells. Carcinogenesis 2018, 39, 601–613. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Lozano-Sánchez, J.; Bastidas-Velez, C.; Llorach-Parés, L.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Joven, J.; Nonell-Canals, A.; Bosch-Barrera, J.; et al. An olive oil phenolic is a new chemotype of mutant isocitrate dehydrogenase 1 (IDH1) inhibitors. Carcinogenesis 2019, 40, 27–40. [Google Scholar] [CrossRef]
- Cuyàs, E.; Castillo, D.; Llorach-Parés, L.; Lozano-Sánchez, J.; Verdura, S.; Nonell-Canals, A.; Brunet, J.; Bosch-Barrera, J.; Joven, J.; Valdés, R.; et al. Computational de-orphanization of the olive oil biophenol oleacein: Discovery of new metabolic and epigenetic targets. Food Chem. Toxicol. 2019, 131, 110529. [Google Scholar] [CrossRef]
- Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2′-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef]
- Tramonti, A.; Cuyàs, E.; Encinar, J.A.; Pietzke, M.; Paone, A.; Verdura, S.; Arbusà, A.; Martin-Castillo, B.; Giardina, G.; Joven, J.; et al. Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers 2021, 13, 4009. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Lozano-Sánchez, J.; Viciano, I.; Llorach-Parés, L.; Nonell-Canals, A.; Bosch-Barrera, J.; Brunet, J.; Segura-Carretero, A.; Sanchez-Martinez, M.; et al. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem. Toxicol. 2019, 128, 35–45. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Pérez-Sánchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Ruiz-Torres, V.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Menendez, J.A. Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals 2021, 14, 559. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DG (n = 9) | TG (n = 25) | PC (n = 40) | LPC (n = 9) | PE (n = 4) | SM (n = 24) | ChoE (n = 13) |
|---|---|---|---|---|---|---|
| DG 34:1 | TG 46:0 | PC 30:0 | LPC 15:0 | PE 32:0 | SM 32:0 | ChoE 16:0 |
| DG 34:2 | TG 46:1 | PC 31:0 | LPC 16:0 | PE 36:4 | SM 32:1 | ChoE 16:1 |
| DG 34:3 | TG 46:2 | PC 32:0 | LPC 16:0e | PE 36:5e | SM 32:2 | ChoE 17:0 |
| DG 36:0 | TG 48:0 | PC 32:1 | LPC 18:0 | PE 38:5e | SM 33:1 | ChoE 17:1 |
| DG 36:1 | TG 48:1 | PC 32:1e | LPC 18:0e | SM 34:1 | ChoE 18:0 | |
| DG 36:2 | TG 48:2 | PC 32:2 | LPC 18:1 | SM 34:2 | ChoE 18:2 | |
| DG 36:3 | TG 48:3 | PC 33:0 | LPC 18:2 | SM 35:0 | ChoE 18:3 | |
| DG 36:4 | TG 50:0 | PC 33:1 | LPC 20:0 | SM 35:1 | ChoE 20:2 | |
| DG 40:4 | TG 50:1 | PC 33:2 | LPC 20:3 | SM 36:0 | ChoE 20:3 | |
| TG 50:2 | PC 34:0 | SM 36:1 | ChoE 20:4 | |||
| TG 50:3 | PC 34:1 | SM 36:2 | ChoE 20:5 | |||
| TG 50:4 | PC 34:1e | SM 38:1 | ChoE 22:4 | |||
| TG 51:2 | PC 34:2 | SM 38:2 | ChoE 22:5 | |||
| TG 52:1 | PC 34:2e | SM 39:1 | ||||
| TG 52:2 | PC 34:3 | SM 40:0 | ||||
| TG 52:3 | PC 34:4 | SM 40:1 | ||||
| TG 52:4 | PC 35:1 | SM 40:2 | ||||
| TG 52:5 | PC 35:2 | SM 41:1 | ||||
| TG 52:6 | PC 35:4 | SM 41:2 | ||||
| TG 54:2 | PC 36:0 | SM 42:1 | ||||
| TG 54:3 | PC 36:1 | SM 42:2 | ||||
| TG 54:4 | PC 36:2 | SM 42:3 | ||||
| TG 54:5 | PC 36:2e | SM 43:1 | ||||
| TG 54:6 | PC 36:3 | SM 43:2 | ||||
| TG 54:7 | PC 36:4 | |||||
| PC 36:5 | ||||||
| PC 36:5e | ||||||
| PC 38:2 | ||||||
| PC 38:3 | ||||||
| PC 38:4 | ||||||
| PC 38:5 | ||||||
| PC 38:5e | ||||||
| PC 38:6 | ||||||
| PC 38:6e | ||||||
| PC 40:4 | ||||||
| PC 40:4e | ||||||
| PC 40:5 | ||||||
| PC 40:6 | ||||||
| PC 42:4e | ||||||
| PC 42:5e |
| ΔG (kcal/mol) | Kd [nM] | Drug | Residues Involved in the Interaction (7KVS.pdb) |
|---|---|---|---|
| −10.323 | 27.1 | silibinin A | TYR 53, PHE 57, ASP 76, ARG 105, ARG 106, PHE 108, GLY 109, PHE 213, PHE 215, PHE 220, ILE 223, THR 224, PRO 227, ILE 230, VAL 240, ALA 370, MET 371, ARG 372, LEU 373, GLU 374, ARG 375, HEME 601 |
| −9.962 | 49.9 | ritonavir | PHE 57, ARG 105, ARG 106, PHE 108, MET 114, SER 119, ILE 120, LEU 210, LEU 211, PHE 213, PHE 241, ILE 300, ILE 301, PHE 304, ALA 305, THR 309, ILE 369, ALA 370, MET 371, ARG 372, LEU 373, GLU 374, HEME 601 |
| −9.876 | 57.6 | lorlatinib | PHE 57, ARG 105, SER 119, LEU 211, PHE 304, GLU 308, THR 309, SER 312, ILE 369, ALA 370, MET 371, LEU 373, LEU 482, HEME 601 |
| −9.651 | 84.3 | silibinin B | PHE 57, ARG 105, ARG 106, PRO 107, PHE 108, SER 119, ILE 301, ALA 305, THR 309, ALA 370, MET 371, ARG 372, LEU 373, GLU 374, HEME 601 |
| Cytochrome P450 Isoforms | |||||
|---|---|---|---|---|---|
| Drug | 1A2 | 2C9 | 2C19 | 2D6 | 3A4 |
| Ritonavir | 0.00 | 0.34 | 0.36 | 0.01 | 0.97 |
| Simvastatin | 0.00 | 0.02 | 0.04 | 0.00 | 0.93 |
| Rosuvastatin | 0.00 | 0.45 | 0.18 | 0.00 | 0.2 |
| Silibinin | 0.00 | 0.02 | 0.04 | 0.00 | 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdura, S.; Encinar, J.A.; Fernández-Arroyo, S.; Joven, J.; Cuyàs, E.; Bosch-Barrera, J.; Menendez, J.A. Silibinin Suppresses the Hyperlipidemic Effects of the ALK-Tyrosine Kinase Inhibitor Lorlatinib in Hepatic Cells. Int. J. Mol. Sci. 2022, 23, 9986. https://doi.org/10.3390/ijms23179986
Verdura S, Encinar JA, Fernández-Arroyo S, Joven J, Cuyàs E, Bosch-Barrera J, Menendez JA. Silibinin Suppresses the Hyperlipidemic Effects of the ALK-Tyrosine Kinase Inhibitor Lorlatinib in Hepatic Cells. International Journal of Molecular Sciences. 2022; 23(17):9986. https://doi.org/10.3390/ijms23179986
Chicago/Turabian StyleVerdura, Sara, José Antonio Encinar, Salvador Fernández-Arroyo, Jorge Joven, Elisabet Cuyàs, Joaquim Bosch-Barrera, and Javier A. Menendez. 2022. "Silibinin Suppresses the Hyperlipidemic Effects of the ALK-Tyrosine Kinase Inhibitor Lorlatinib in Hepatic Cells" International Journal of Molecular Sciences 23, no. 17: 9986. https://doi.org/10.3390/ijms23179986
APA StyleVerdura, S., Encinar, J. A., Fernández-Arroyo, S., Joven, J., Cuyàs, E., Bosch-Barrera, J., & Menendez, J. A. (2022). Silibinin Suppresses the Hyperlipidemic Effects of the ALK-Tyrosine Kinase Inhibitor Lorlatinib in Hepatic Cells. International Journal of Molecular Sciences, 23(17), 9986. https://doi.org/10.3390/ijms23179986