
Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity

 ,
,
Abstract
:1. Introduction
2. Macrophage Polarization and Cellular Energy Metabolism
2.1. M1 Macrophages
2.2. M2 Macrophages
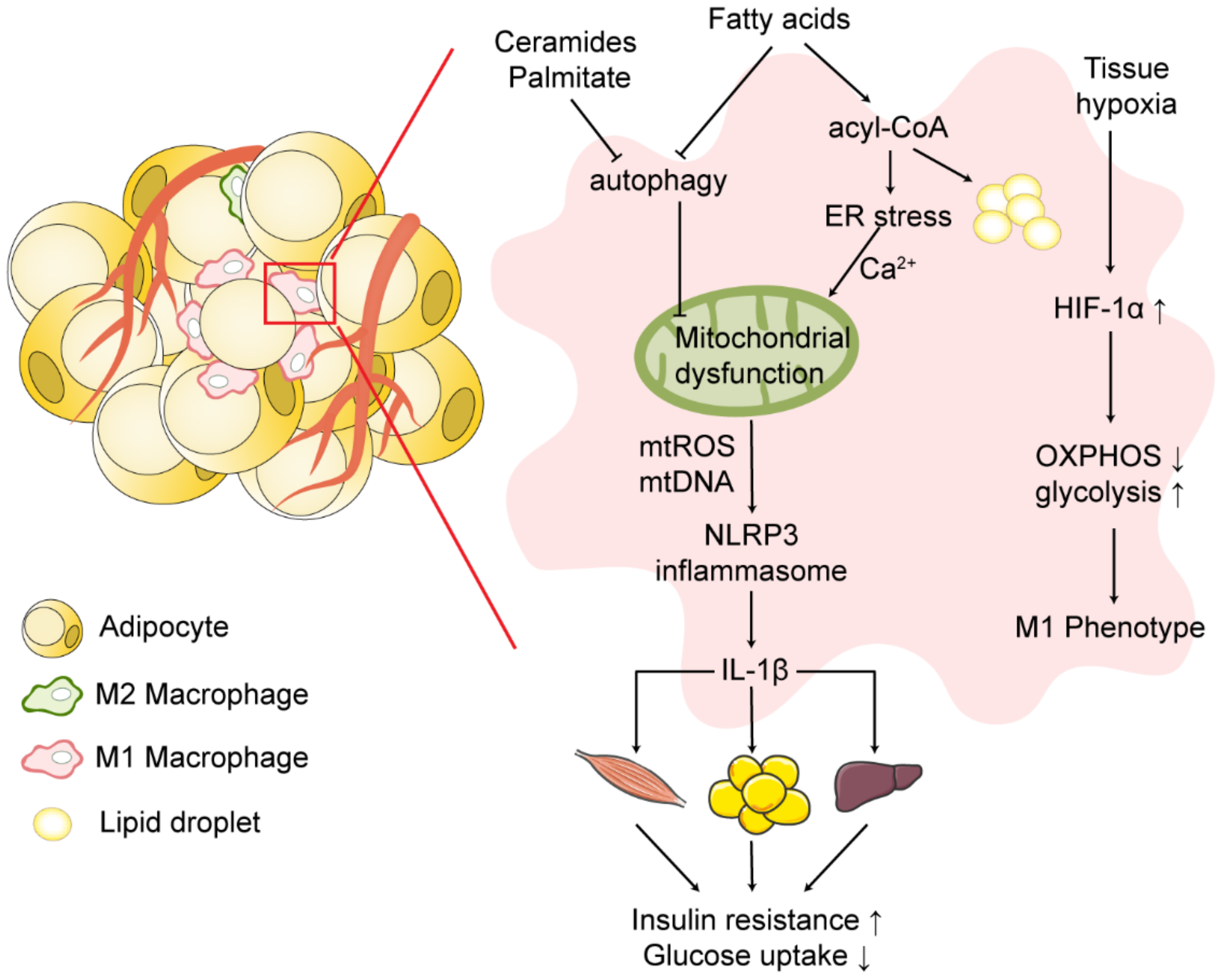
3. Mitochondrial Dysfunction of Macrophages in Obese Adipose Tissue
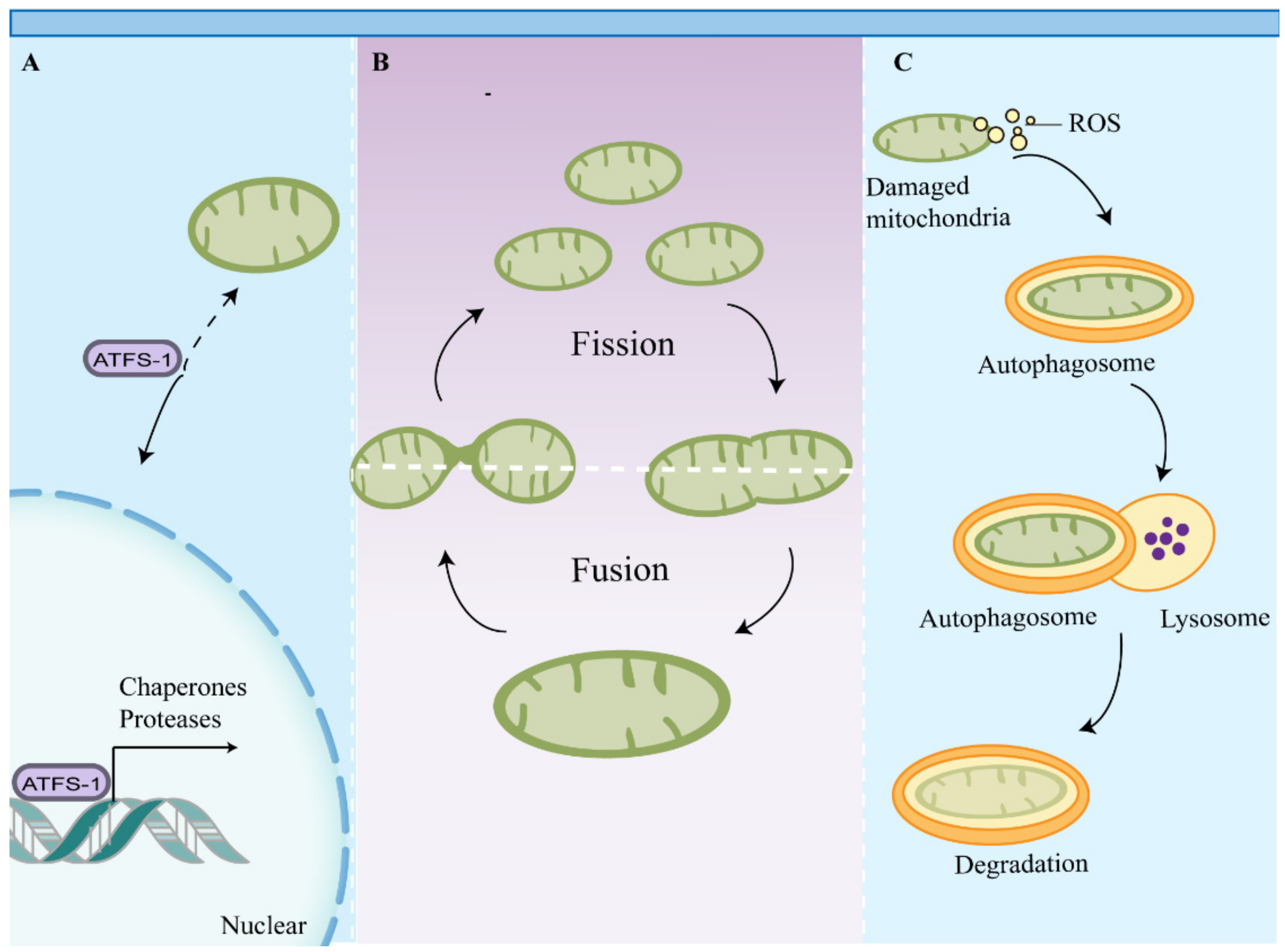
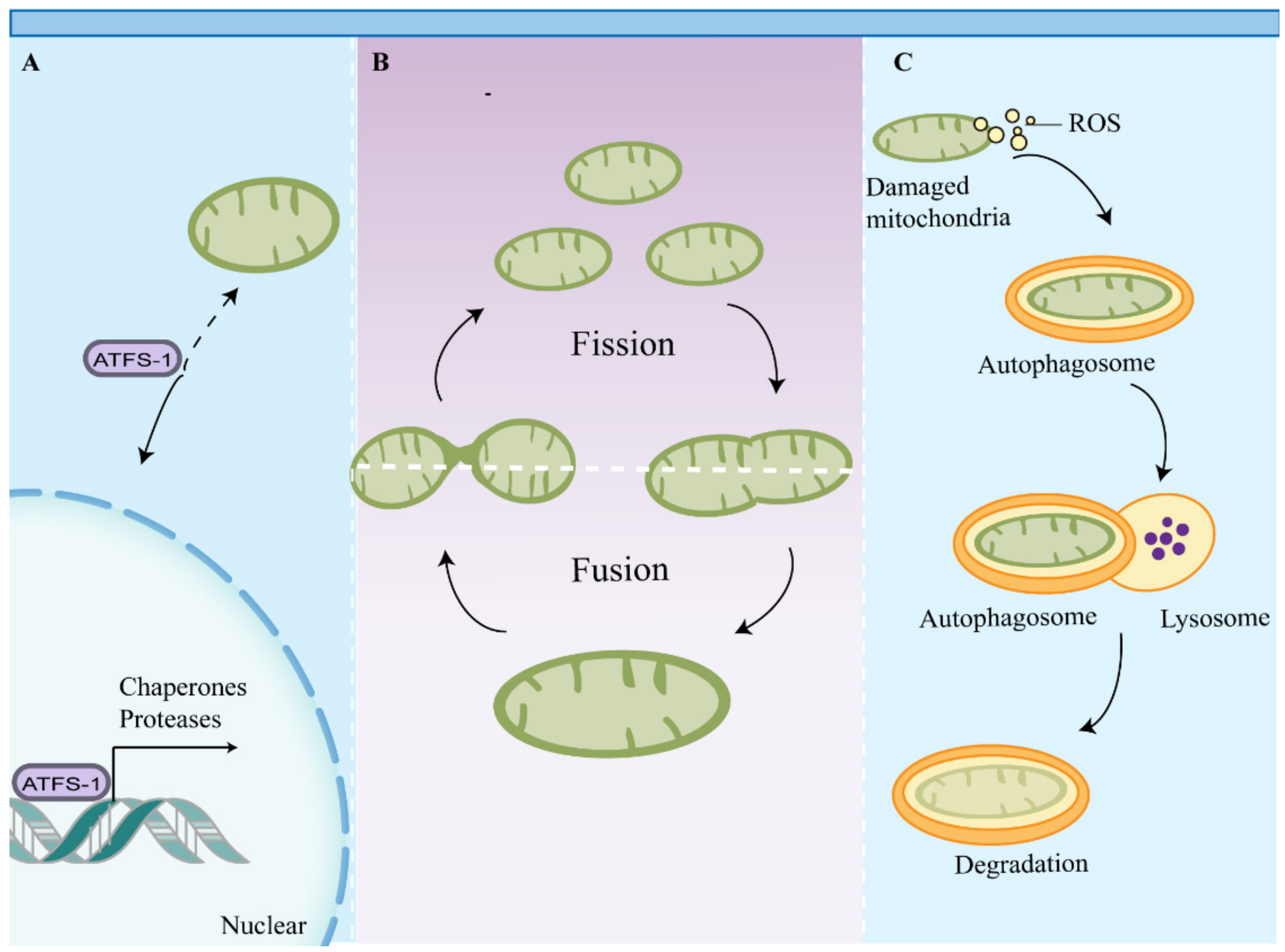
4. Impaired Mitochondrial Quality Control Leads to the Accumulation of Dysfunctional Mitochondria in Macrophages
5. Mitochondrial Dysfunction Drives NLRP3 Inflammasome Activation
6. Mitochondrial Dysfunction Impairs Macrophage Function and Plasticity
7. Adipose Tissue Macrophages Link Adipose Tissue Inflammation to Insulin Resistance
8. Mitochondrially Targeted Treatment Strategies
8.1. Lifestyle
8.2. Pharmacological Therapy

{kind=link}
{kind=link}
| Substance | Pathway or Category | Effects on Mitochondria | References |
|---|---|---|---|
| TZDs | PPARs agonistic | Enhanced fatty acid oxidation | [161] |
| Metformin | Complex I inhibition | Inhibited respiratory chain | [162] |
| Pioglitazone | PPAR-γ/PGC-1α Pathway | Improved mitochondrial biogenesis and dynamics | [163] |
| Resveratrol | SIRT1-PGC-1α axis | Induced mitochondrial biogenesis | [164] |
| MitoQ | Nrf2/PINK1 pathway | Restored mitophagy | [159] |
| Mdivi1 | Inhibitor of Drp1 | Inhibited mitochondrial fission | [165] |
| Dynasore | Inhibitor of Drp1 | Inhibited mitochondrial fission | [166] |
| Empagliflozin | AMPK/SP1/PGAM5 pathway | Inhibited mitochondrial fission | [167] |
| rPGRN | PGRN- SIRT1-PGC-1α/FoxO1 pathway | Enhanced mitochondrial biogenesis and mitophagy | [160] |
8.3. Novel Mitochondria-Targeted Therapeutic Approaches
9. Conclusions and Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Gericke, M.; Weyer, U.; Braune, J.; Bechmann, I.; Eilers, J. A method for long-term live imaging of tissue macrophages in adipose tissue explants. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1023–E1033. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DeYoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased Inflammatory Properties of Adipose Tissue Macrophages Recruited During Diet-Induced Obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic Switching of Adipose Tissue Macrophages with Obesity Is Generated by Spatiotemporal Differences in Macrophage Subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Zhang, X.; Horng, T. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021, 297, 100904. [Google Scholar] [CrossRef]
- Wu, K.K.-L.; Cheung, S.W.-M.; Cheng, K.K.-Y. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 4184. [Google Scholar] [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef]
- Kim, Y.; Wang, W.; Okla, M.; Kang, I.; Moreau, R.; Chung, S. Suppression of NLRP3 inflammasome by γ-tocotrienol ameliorates type 2 diabetes. J. Lipid Res. 2016, 57, 66–76. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 65–80, Erratum in Cell Metab. 2015, 21, 347. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; O’Neill, L.A.J. The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays 2013, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Enagy, C.; Ehaschemi, A. Time and Demand are Two Critical Dimensions of Immunometabolism: The Process of Macrophage Activation and the Pentose Phosphate Pathway. Front. Immunol. 2015, 6, 164. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.V.; Saraber, D.L. Metabolic regulation of macrophages in tissues. Cell. Immunol. 2018, 330, 54–59. [Google Scholar] [CrossRef]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef]
- Nomura, M.; Liu, J.; Rovira, I.I.; Gonzalez-Hurtado, E.; Lee, J.; Wolfgang, M.J.; Finkel, T. Fatty acid oxidation in macrophage polarization. Nat. Immunol. 2016, 17, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Boutens, L.; Stienstra, R. Adipose tissue macrophages: Going off track during obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef]
- Shapiro, H.; Pecht, T.; Shaco-Levy, R.; Harman-Boehm, I.; Kirshtein, B.; Kuperman, Y.; Chen, A.; Blüher, M.; Shai, I.; Rudich, A. Adipose Tissue Foam Cells Are Present in Human Obesity. J. Clin. Endocrinol. Metab. 2013, 98, 1173–1181. [Google Scholar] [CrossRef]
- Robblee, M.M.; Kim, C.C.; Abate, J.P.; Valdearcos, M.; Sandlund, K.L.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.N.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of Lysosomal Biogenesis in Atherosclerotic Macrophages Can Rescue Lipid-Induced Lysosomal Dysfunction and Downstream Sequelae. Arter. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e11. [Google Scholar] [CrossRef]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining Ancient Organelles: Mitochondrial biogenesis and maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef]
- Stuart, J.A.; Brown, M.F. Mitochondrial DNA maintenance and bioenergetics. Biochim. Biophys. Acta 2006, 1757, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Wang, B.; Wood, I.S. Hypoxia in adipose tissue: A basis for the dysregulation of tissue function in obesity? Br. J. Nutr. 2008, 100, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose Tissue Hypoxia in Obesity and Its Impact on Adipocytokine Dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Gao, Z.; Yin, J.; He, Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1118–E1128. [Google Scholar] [CrossRef]
- Catrysse, L.; van Loo, G. Adipose tissue macrophages and their polarization in health and obesity. Cell. Immunol. 2018, 330, 114–119. [Google Scholar] [CrossRef]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef]
- Fujisaka, S.; Usui, I.; Ikutani, M.; Aminuddin, A.; Takikawa, A.; Tsuneyama, K.; Mahmood, A.; Goda, N.; Nagai, Y.; Takatsu, K.; et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia 2013, 56, 1403–1412. [Google Scholar] [CrossRef]
- Takikawa, A.; Mahmood, A.; Nawaz, A.; Kado, T.; Okabe, K.; Yamamoto, S.; Aminuddin, A.; Senda, S.; Tsuneyama, K.; Ikutani, M.; et al. HIF-1α in Myeloid Cells Promotes Adipose Tissue Remodeling Toward Insulin Resistance. Diabetes 2016, 65, 3649–3659. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Maceyka, M.; Chen, Q. Targeting ER stress and calpain activation to reverse age-dependent mitochondrial damage in the heart. Mech. Ageing Dev. 2020, 192, 111380. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Nguyen, K.Y.; Jurczak, M.J.; Christian, B.E.; Shulman, G.I.; Shadel, G.S.; Dixit, V.D. Macrophage-specific de Novo Synthesis of Ceramide Is Dispensable for Inflammasome-driven Inflammation and Insulin Resistance in Obesity. J. Biol. Chem. 2015, 290, 29402–29413. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D.; Brüne, B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1796–1807. [Google Scholar] [CrossRef]
- Haneklaus, M.; O’Neill, L. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Zezina, E.; Snodgrass, R.G.; Schreiber, Y.; Zukunft, S.; Schürmann, C.; zu Heringdorf, D.M.; Geisslinger, G.; Fleming, I.; Brandes, R.P.; Brüne, B.; et al. Mitochondrial fragmentation in human macrophages attenuates palmitate-induced inflammatory responses. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2018, 1863, 433–446. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Moehlman, A.T.; Youle, R.J. Mitochondrial Quality Control and Restraining Innate Immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Weidberg, H.; Amon, A. MitoCPR—A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018, 360, eaan4146. [Google Scholar] [CrossRef]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 2015, 524, 485–488. [Google Scholar] [CrossRef]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.; et al. Altered Autophagy in Human Adipose Tissues in Obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef]
- Kraunsøe, R.; Boushel, R.; Hansen, C.N.; Schjerling, P.; Qvortrup, K.; Støckel, M.; Mikines, K.J.; Dela, F. Mitochondrial respiration in subcutaneous and visceral adipose tissue from patients with morbid obesity. J. Physiol. 2010, 588, 2023–2032. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell. Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Kajimura, S. Mitochondrial homeostasis in adipose tissue remodeling. Sci. Signal. 2017, 10, eaai9248. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Kanneganti, T.-D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Yu, J.-W.; Lee, M.-S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharm. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225, Erratum in Nature 2011, 475, 122. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Fernández-Ayala, D.J.M.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.; Lucas, C.; Rossi, A.G.; Ravichandran, K. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Hardy, O.T.; Perugini, R.A.; Nicoloro, S.M.; Gallagher-Dorval, K.; Puri, V.; Straubhaar, J.; Czech, M.P. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg. Obes. Relat. Dis. 2011, 7, 60–67. [Google Scholar] [CrossRef]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P.S. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef]
- McLaughlin, T.; Ackerman, S.E.; Shen, L.; Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Investig. 2017, 127, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, A.; Naffah de Souza, C.; Camara, N.O.S.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2015, 6, 637. [Google Scholar] [CrossRef] [PubMed]
- Boulenouar, S.; Michelet, X.; Duquette, D.; Alvarez, D.; Hogan, A.E.; Dold, C.; O’Connor, D.; Stutte, S.; Tavakkoli, A.; Winters, D.; et al. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 2017, 46, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Serbulea, V.; Upchurch, C.M.; Schappe, M.S.; Voigt, P.; DeWeese, D.E.; Desai, B.N.; Meher, A.K.; Leitinger, N. Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E6254–E6263. [Google Scholar] [CrossRef]
- Watanabe, R.; Hilhorst, M.; Zhang, H.; Zeisbrich, M.; Berry, G.J.; Wallis, B.B.; Harrison, D.G.; Giacomini, J.C.; Goronzy, J.J.; Weyand, C.M. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight 2018, 3, e123047. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef]
- Cosentino, C.; Regazzi, R. Crosstalk between Macrophages and Pancreatic β-Cells in Islet Development, Homeostasis and Disease. Int. J. Mol. Sci. 2021, 22, 1765. [Google Scholar] [CrossRef]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.-Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal Visceral and Subcutaneous Adipose Tissue Compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; Joosten, L.A.B.; van Velzen, J.F.; Hijmans, A.; Pol, J.A.; van der Vliet, J.A.; Netea, M.G.; Tack, C.J.; et al. The Inflammasome and Caspase-1 Activation: A New Mechanism Underlying Increased Inflammatory Activity in Human Visceral Adipose Tissue. Endocrinology 2011, 152, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Bar Hen, A.; Bedossa, P.; et al. Increased Infiltration of Macrophages in Omental Adipose Tissue Is Associated with Marked Hepatic Lesions in Morbid Human Obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T., Jr.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, R.W.; White, A.E.; Metcalf, M.D.; Olivas, A.S.; Mitra, P.; Larison, W.G.; Cheang, E.C.; Varlamov, O.; Corless, C.L.; Roberts, C.T.; et al. Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells. Diabetologia 2011, 54, 1480–1490. [Google Scholar] [CrossRef]
- Spoto, B.; Di Betta, E.; Mattace-Raso, F.; Sijbrands, E.; Vilardi, A.; Parlongo, R.; Pizzini, P.; Pisano, A.; Vermi, W.; Testa, A.; et al. Pro- and anti-inflammatory cytokine gene expression in subcutaneous and visceral fat in severe obesity. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1137–1143. [Google Scholar] [CrossRef]
- Lambers, S.; Van Laethem, C.; Van Acker, K.; Calders, P. Influence of combined exercise training on indices of obesity, diabetes and cardiovascular risk in type 2 diabetes patients. Clin. Rehabil. 2008, 22, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.S.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef]
- Meex, R.C.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.-P.; Schrauwen, P.; Hesselink, M.K. Restoration of Muscle Mitochondrial Function and Metabolic Flexibility in Type 2 Diabetes by Exercise Training Is Paralleled by Increased Myocellular Fat Storage and Improved Insulin Sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Russell, A.P.; Foletta, V.C.; Snow, R.J.; Wadley, G.D. Skeletal muscle mitochondria: A major player in exercise, health and disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1276–1284. [Google Scholar] [CrossRef]
- Barbieri, E.; Agostini, D.; Polidori, E.; Potenza, L.; Guescini, M.; Lucertini, F.; Annibalini, G.; Stocchi, L.; De Santi, M.; Stocchi, V. The Pleiotropic Effect of Physical Exercise on Mitochondrial Dynamics in Aging Skeletal Muscle. Oxid. Med. Cell. Longev. 2015, 2015, 917085. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The Retardation of Aging in Mice by Dietary Restriction: Longevity, Cancer, Immunity and Lifetime Energy Intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef]
- Omodei, D.; Fontana, L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011, 585, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; the CALERIE Pennington Team. Calorie Restriction Increases Muscle Mitochondrial Biogenesis in Healthy Humans. PLOS Med. 2007, 4, e76. [Google Scholar] [CrossRef] [PubMed]
- Wierman, M.B.; Maqani, N.; Strickler, E.; Li, M.; Smith, J.S. Caloric Restriction Extends Yeast Chronological Life Span by Optimizing the Snf1 (AMPK) Signaling Pathway. Mol. Cell. Biol. 2017, 37, e00562-16. [Google Scholar] [CrossRef] [PubMed]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896.e5. [Google Scholar] [CrossRef]
- Stenesen, D.; Suh, J.M.; Seo, J.; Yu, K.; Lee, K.-S.; Kim, J.-S.; Min, K.-J.; Graff, J.M. Adenosine Nucleotide Biosynthesis and AMPK Regulate Adult Life Span and Mediate the Longevity Benefit of Caloric Restriction in Flies. Cell Metab. 2013, 17, 101–112. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef]
- Kemnitz, J.W.; Roecker, E.B.; Weindruch, R.; Elson, D.F.; Baum, S.T.; Bergman, R.N. Dietary restriction increases insulin sensitivity and lowers blood glucose in rhesus monkeys. Am. J. Physiol. Metab. 1994, 266, E540–E547. [Google Scholar] [CrossRef]
- Zainal, T.A.; Oberley, T.D.; Allison, D.B.; Szweda, L.I.; Weindruch, R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. FASEB J. 2000, 14, 1825–1836. [Google Scholar] [CrossRef]
- Sharma, N.; Arias, E.B.; Bhat, A.D.; Sequea, D.A.; Ho, S.; Croff, K.K.; Sajan, M.P.; Farese, R.V.; Cartee, G.D. Mechanisms for increased insulin-stimulated Akt phosphorylation and glucose uptake in fast- and slow-twitch skeletal muscles of calorie-restricted rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E966–E978. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Dirks, A.J.; Leeuwenburgh, C. Caloric restriction in humans: Potential pitfalls and health concerns. Mech. Ageing Dev. 2006, 127, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Wiley, S.E.; Rogers, G.W.; Andreyev, A.Y.; Petrosyan, S.; Loviscach, M.; Wall, E.A.; Yadava, N.; Heuck, A.P.; Ferrick, D.A.; et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA 2013, 110, 5422–5427. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma; controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. Cell. Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef]
- Fridlyand, L.E.; Philipson, L.H. Reactive Species, Cellular Repair and Risk Factors in the Onset of Type 2 Diabetes Mellitus: Review and Hypothesis. Curr. Diabetes Rev. 2006, 2, 241–259. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective Targeting of a Redox-active Ubiquinone to Mitochondria within Cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Murphy, M.P. Mitochondria-targeted antioxidants as therapies. Discov. Med. 2011, 11, 106–114. [Google Scholar]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hao, C.; Liu, X.; Han, G.; Yin, J.; Zou, Z.; Zhou, J.; Xu, C. MitoQ protects against liver injury induced by severe burn plus delayed resuscitation by suppressing the mtDNA-NLRP3 axis. Int. Immunopharmacol. 2020, 80, 106189. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A–mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. A biochemical approach to the problem of aging: “Megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry 2007, 72, 1385–1396. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin–Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Xu, L.; Li, X.; Sun, H.; Xue, M.; Li, T.; Yu, X.; Sun, B.; Chen, L. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism 2020, 111, 154334. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Colca, J.R.; McDonald, W.G.; Kletzien, R.F. Mitochondrial target of thiazolidinediones. Diabetes Obes. Metab. 2014, 16, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Iannantuoni, F.; Gruevska, A.; Muntane, J.; Rocha, M.; Victor, V.M. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020, 34, 101517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmacol. 2021, 12, 658362. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.-J.; Wang, C.-J.; He, Y.; Zhou, Y.-L.; Peng, X.; Liu, S.-K. Resveratrol alleviates diabetic cardiomyopathy in rats by improving mitochondrial function through PGC-1α deacetylation. Acta Pharmacol. Sin. 2018, 39, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Weng, S.-W.; Chang, Y.-H.; Su, Y.-J.; Chang, C.-M.; Tsai, C.-J.; Shen, F.-C.; Chuang, J.-H.; Lin, T.-K.; Liou, C.-W.; et al. The Causal Role of Mitochondrial Dynamics in Regulating Insulin Resistance in Diabetes: Link through Mitochondrial Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2018, 2018, 7514383. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.-T.; Shaw, R.M.; Zhu, J. Dynasore Protects Mitochondria and Improves Cardiac Lusitropy in Langendorff Perfused Mouse Heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef]
- Dai, W.; Lu, H.; Chen, Y.; Yang, D.; Sun, L.; He, L. The Loss of Mitochondrial Quality Control in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2021, 9, 706832. [Google Scholar] [CrossRef]
- Keijer, J.; van Schothorst, E. Adipose tissue failure and mitochondria as a possible target for improvement by bioactive food components. Curr. Opin. Lipidol. 2008, 19, 4–10. [Google Scholar] [CrossRef]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce β-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Dos Santos, T.W.; Pereira, Q.C.; Teixeira, L.; Gambero, A.; Villena, J.A.; Ribeiro, M.L. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19, 2757. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Huang, C.; Wang, C.; Zheng, J.; Zhang, P.; Xu, Y.; Chen, H.; Shen, W. Ginsenoside Rg3 improves cardiac mitochondrial population quality: Mimetic exercise training. Biochem. Biophys. Res. Commun. 2013, 441, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-K.; Shim, J.; Cho, A.-R.; Cho, M.-R.; Lee, Y.-J. Korean Red Ginseng Protects Against Mitochondrial Damage and Intracellular Inflammation in an Animal Model of Type 2 Diabetes Mellitus. J. Med. Food 2018, 21, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Soto-Urquieta, M.G.; López-Briones, S.; Pérez-Vázquez, V.; Saavedra-Molina, A.; A González-Hernández, G.; Ramírez-Emiliano, J. Curcumin restores mitochondrial functions and decreases lipid peroxidation in liver and kidneys of diabetic db/db mice. Biol. Res. 2014, 47, 74. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, P.; Luo, Y.; Wu, C.; Liu, Y.; Qin, X.; Huang, X.; Sun, C. Salidroside stimulates the Sirt1/PGC-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine 2019, 54, 240–247. [Google Scholar] [CrossRef]
- Tong, Y.; Chuan, J.; Bai, L.; Shi, J.; Zhong, L.; Duan, X.; Zhu, Y. The protective effect of shikonin on renal tubular epithelial cell injury induced by high glucose. Biomed. Pharmacother. 2018, 98, 701–708. [Google Scholar] [CrossRef]
- Rashedinia, M.; Khoshnoud, M.J.; Fahlyan, B.K.; Hashemi, S.-S.; Alimohammadi, M.; Sabahi, Z. Syringic Acid: A Potential Natural Compound for the Management of Renal Oxidative Stress and Mitochondrial Biogenesis in Diabetic Rats. Curr. Drug Discov. Technol. 2021, 18, 405–413. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, M.; Zhao, Y.; Gong, J.; Su, H.; Yuan, F.; Fang, K.; Yuan, X.; Yu, X.; Dong, H.; et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br. J. Pharmacol. 2019, 177, 3646–3661. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Park, A.; Oh, M.; Lee, S.; Oh, K.-J.; Lee, E.-W.; Lee, S.; Bae, K.-H.; Han, B.; Kim, W. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef]
- Schmitz, J.; Evers, N.; Awazawa, M.; Nicholls, H.; Brönneke, H.; Dietrich, A.; Mauer, J.; Blüher, M.; Brüning, J. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol. Metab. 2016, 5, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Ley, S.H.; Manson, J.E.; Willett, W.; Satija, A.; Hu, F.B.; Stokes, A.C. Weight History and All-Cause and Cause-Specific Mortality in Three Prospective Cohort Studies. Ann. Intern. Med. 2017, 166, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Bigornia, S.J.; Farb, M.G.; Mott, M.M.; Hess, D.; Carmine, B.; Fiscale, A.; Joseph, L.; Apovian, C.; Gokce, N. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr. Diabetes 2012, 2, e30. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. https://doi.org/10.3390/ijms23169252
Xu L, Yan X, Zhao Y, Wang J, Liu B, Yu S, Fu J, Liu Y, Su J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. International Journal of Molecular Sciences. 2022; 23(16):9252. https://doi.org/10.3390/ijms23169252
Chicago/Turabian StyleXu, Long, Xiaoyu Yan, Yuanxin Zhao, Jian Wang, Buhan Liu, Sihang Yu, Jiaying Fu, Yanan Liu, and Jing Su. 2022. "Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity" International Journal of Molecular Sciences 23, no. 16: 9252. https://doi.org/10.3390/ijms23169252
APA StyleXu, L., Yan, X., Zhao, Y., Wang, J., Liu, B., Yu, S., Fu, J., Liu, Y., & Su, J. (2022). Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. International Journal of Molecular Sciences, 23(16), 9252. https://doi.org/10.3390/ijms23169252