
Feature Selection and Molecular Classification of Cancer Phenotypes: A Comparative Study




Abstract
1. Introduction
2. Results
2.1. Predictive Performance
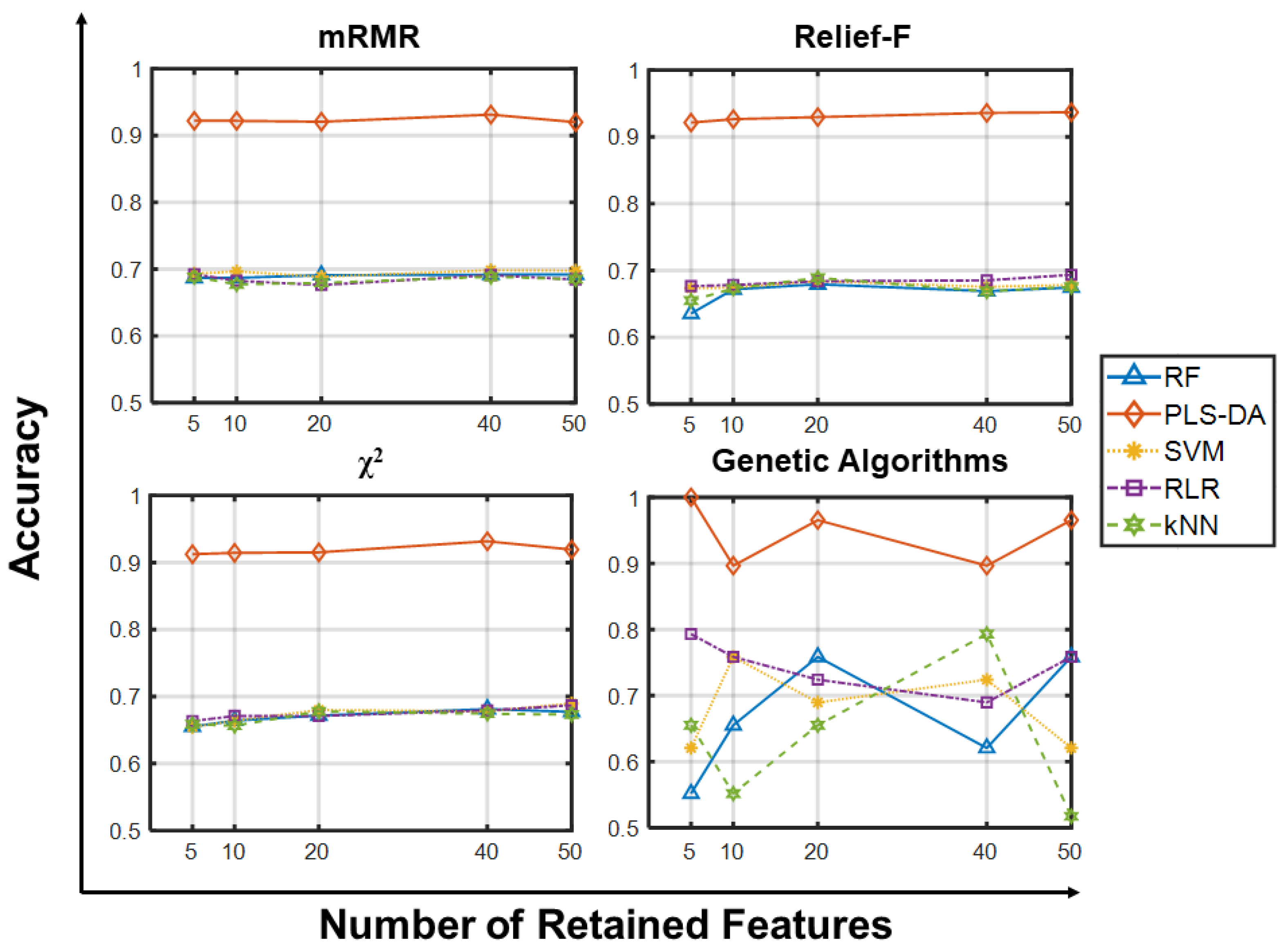
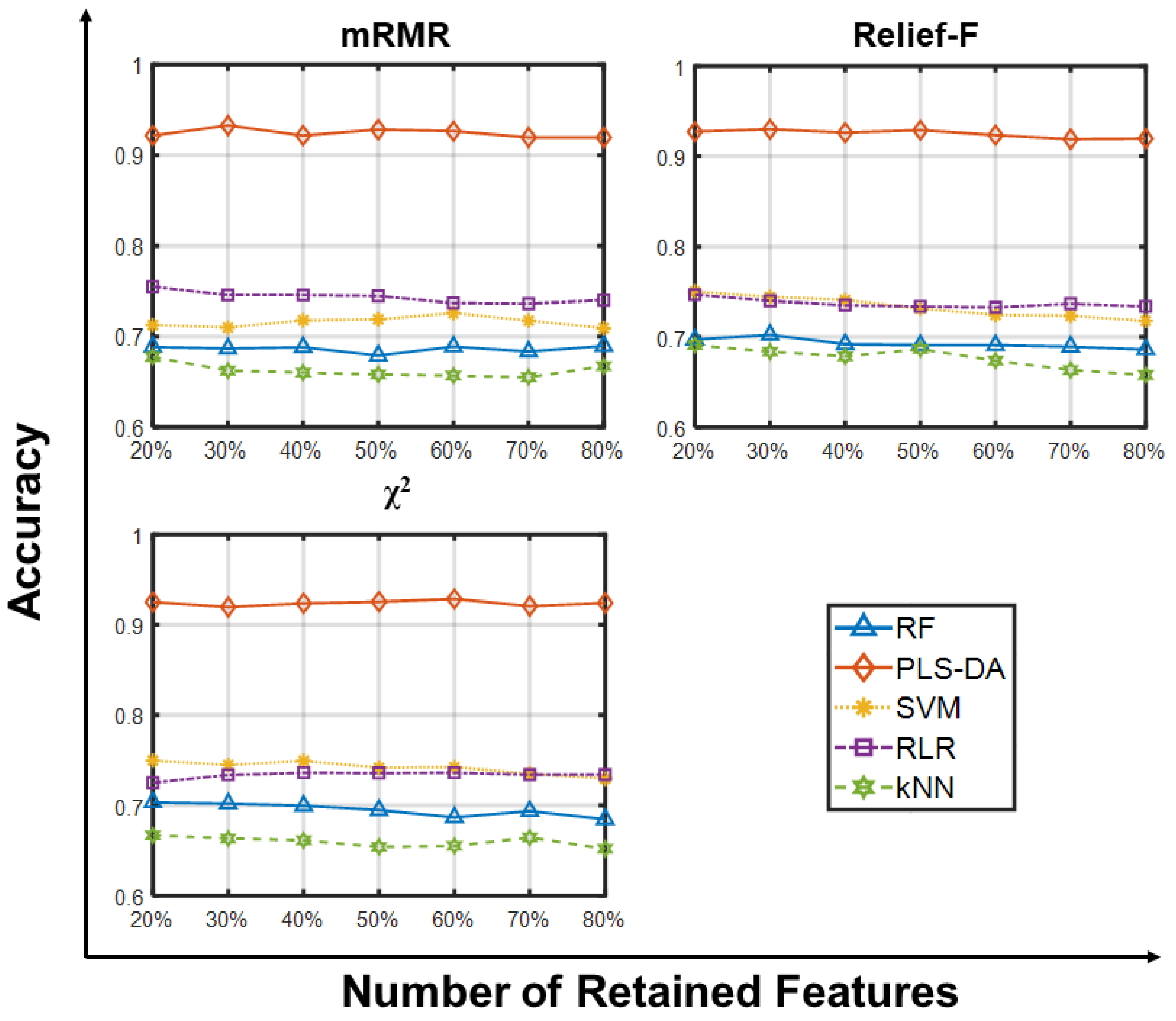
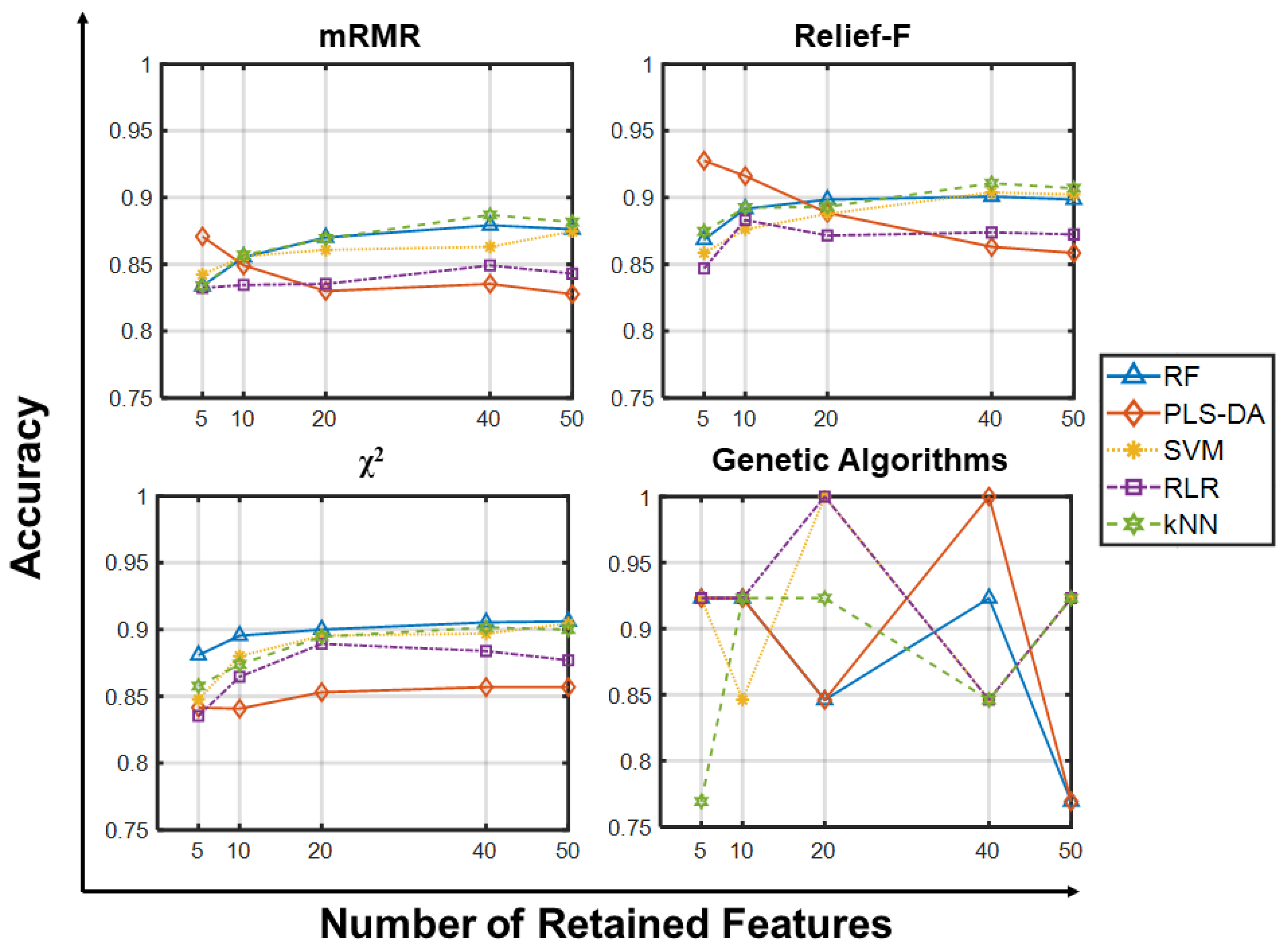
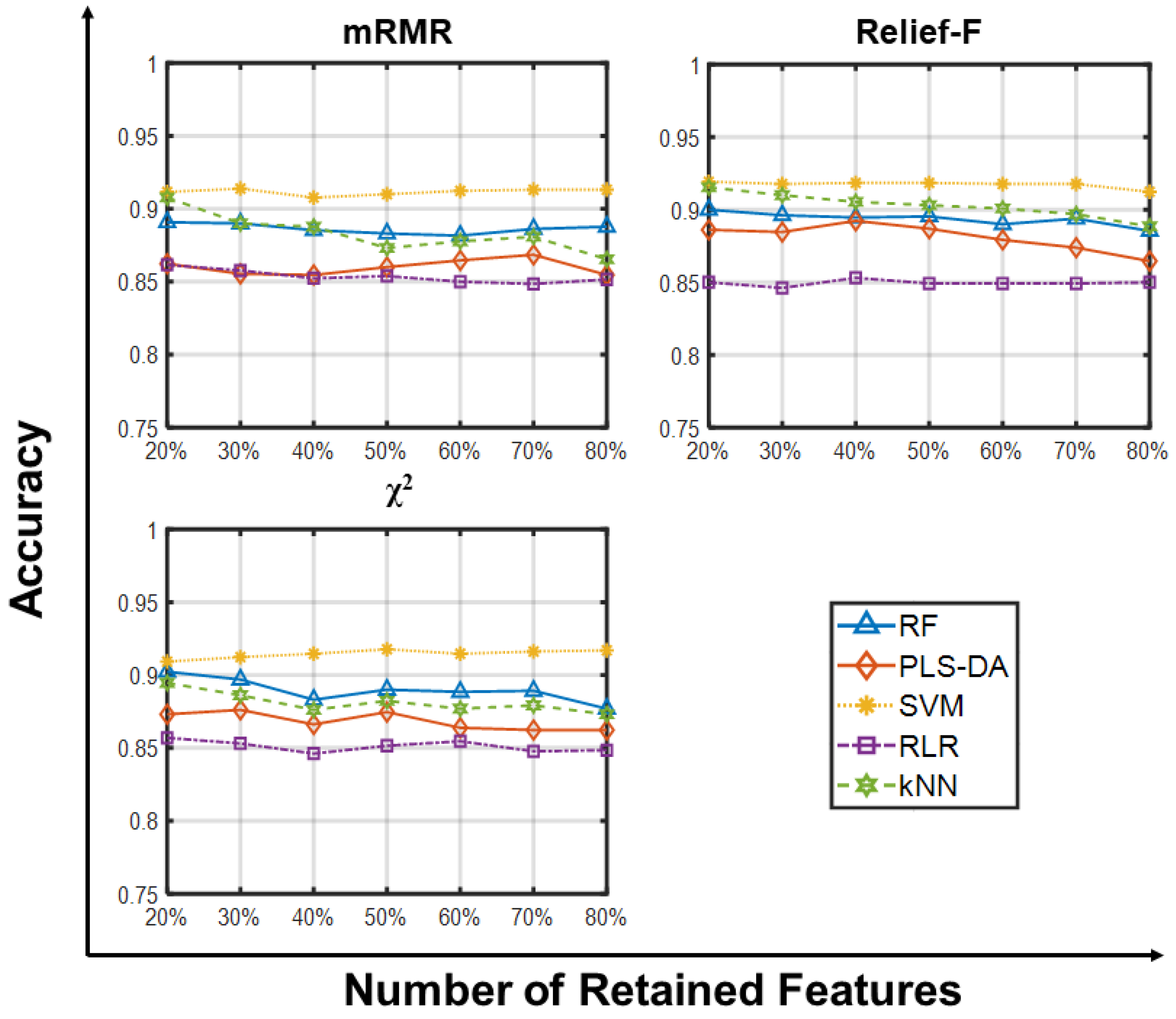
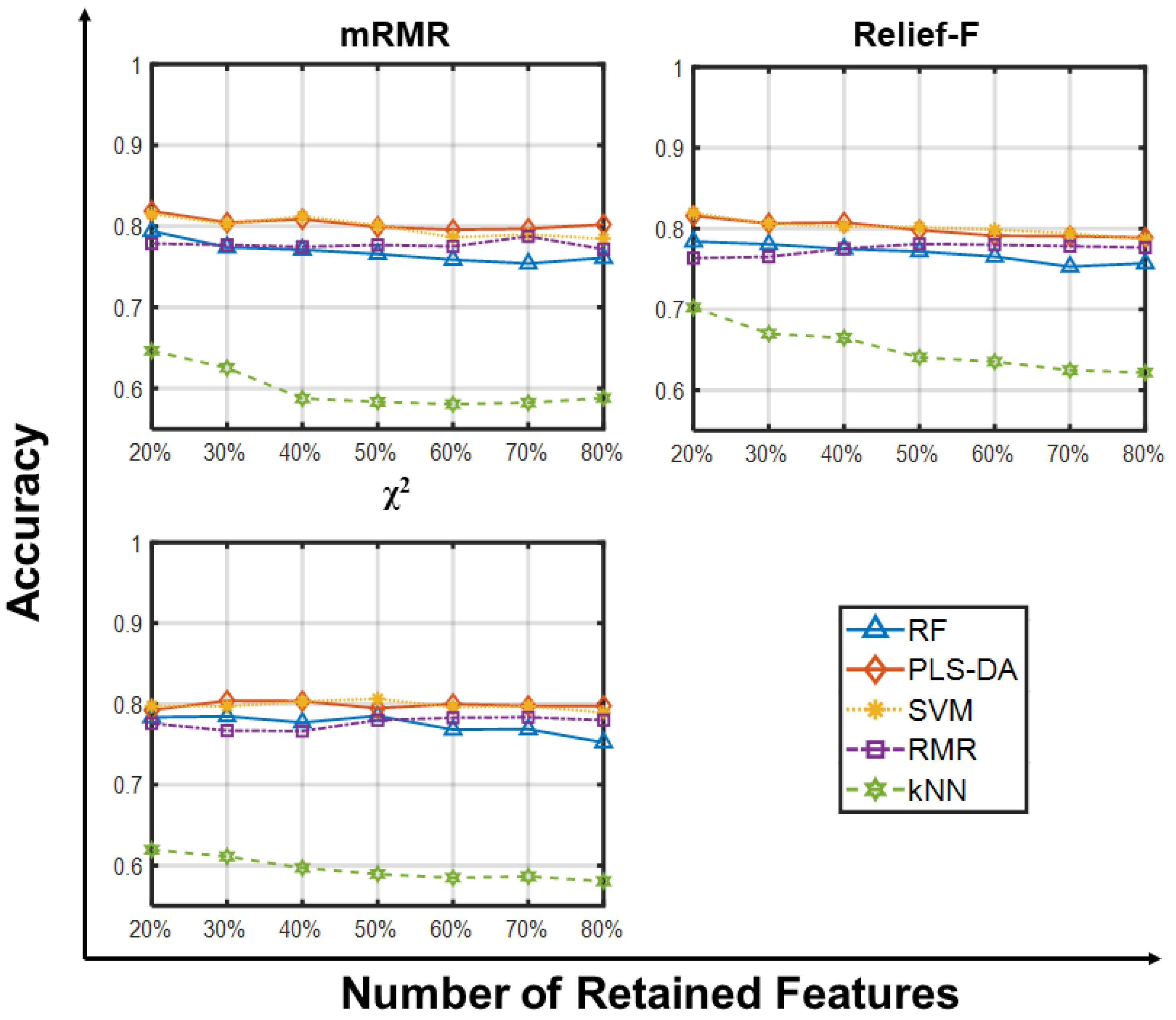
2.1.1. Accuracy Analysis
2.1.2. Other Metrics Analysis
2.1.3. Comparison with Literature Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Accuracy (%) | Feature Selectors | Classification Learning Algorithms | Reference |
|---|---|---|---|---|
| SMK_CAN_187 | 47–83% |
|
| [4] |
| 59–71% |
|
| [28] | |
| 66–71% |
|
| [16] | |
| GLI_85 | 71–85% |
|
| [16] |
| 78–92% |
|
| [28] | |
| 72–80% |
|
| [32] | |
| CLL_SUB_111 | 60–80% |
|
| [16,33] |
2.1.4. Comparison with Variance-Based Unsupervised Feature Selection
2.1.5. Runtime Comparisons
3. Discussion
4. Materials and Methods
4.1. Classification Learning Algorithms
- RF: Random Forests (RF) consist of a combination of a large number of tree predictors, T, each voting for a class, [37]. A bootstrap sample of the training set and a random selection of the input features generate each tree using both bagging [39] and random feature selection. Each decision tree gives a class prediction based on the samples features and gets a vote; the class with the most votes is the forest prediction. RF are not prone to overfitting, even when employing a large number of features, require minimal data cleaning and are effective in variance reduction, while exhibiting good interpretability [40].
- PLS-DA: Partial Least Squares-Discriminant Analysis (PLS-DA) is based on the PLS regression algorithm [41] where the dependent variable represents class membership. The method constructs A latent variables, i.e., linear combinations of the original features, by maximizing their covariance with the target classes. The number of latent variables A, accounting for the dimensionality of the projected space, is the hyperparameter to be optimized in the training phase. Being inherently based on a dimensionality reduction step, PLS-DA is fit for high-dimensional data as it allows locate and emphasize group structures when discrimination is the goal and reduction is needed [42]. Last, the interpretation of PLS-DA models in terms of the contribution of each feature to class discrimination is eased by the analysis of weights (W) and loadings (P) [41], making PLS-DA a versatile tool for predictive and descriptive modelling in biomedical applications and diagnostics [42,43].
- SVM: Support Vector Machines (SVM) [23] is a popular tool for binary classification. SVM seeks the decision boundary that separates all data points of the two classes, i.e., the one with the largest margin between the two classes. The training data is a set of observation-label pairs, and the equation of the hyperplane separating the classes is the solution of an optimization problem. The optimality is influenced only by points that are closer to the decision boundary and thus more difficult to classify. SVM is effective and memory efficient in spanning high-dimensional spaces, as it uses only the support vectors (a subset of the training points) in the decision functions, favoring its application in many areas of bioinformatics [23].
- RLR: Regularized Logistic Regression (RLR) measures the relationship between a categorical class label and one or more features, by estimating probabilities using a logit link function [44]. Here we employed a L1-regularization, robust to irrelevant features and noise in the data [45], and fitted the penalized maximum-likelihood coefficients by solving the optimization problem in (A8) (see Appendix A.1). When generalizing to the multiclass case M > 2, we employed multinomial logistic regression conceived as M–1 independent binary logistic models, where the predicted class is the one with the highest score. Additional information is reported in Appendix A.1. Simple to perform, logistic regression is particularly useful when it comes to model categorical variables, commonly used in biomedical data to encode a set of discrete states (as in the present work). RLR also allows predicting class-associated probability [46] and further enhances model interpretability, particularly favoring applications in high-dimensional domains [44].
- kNN: k-Nearest Neighbors (kNN) is an instance-based method [47]. Given a matrix X of n observations and a distance function, a kNN search finds the k observations in X that are closest to a (or a set of) query samples. The number of nearest neighbors, k, is the main hyperparameter to tune. In general, small values of k lead to classifiers with weak generalization abilities, while large k’s increase the computational demand. kNN assigns labels to new unknown samples using labels of the k most similar samples in the training set, based on their distance to observations (Euclidean in this work). Despite its simplicity, kNN is a suitable choice even when data have nonlinear relationships, making it a benchmark learning rule to draw comparisons with other classification techniques [48].
4.2. Feature Selectors
4.2.1. Filters
- mRMR: the minimum redundancy maximum relevance (mRMR) selects features that are minimally redundant (maximally dissimilar to each other), while being maximally relevant to the target classes [50]. mRMR requires binning, and bins reflect the number of values assumed by each gene. Redundancy and relevance are quantified using the pairwise mutual information of features and mutual information of feature and target classes, respectively [22]. Relevant features are necessary to build optimal subsets, given their strong correlation with the target classes, and the mRMR algorithm performs a sequential selection with a forward addition scheme. At each iteration, the feature that is mostly relevant to the target and least redundant compared to the already selected ones is added [20].
- Relief-F: Relief-F ranks the most informative features using the concept of nearest neighbors by weighting their ability to discriminate observations under different classes using distance-based criteria functions [22]. Features assigning different values to neighbors of different classes are rewarded, while those that give different values to neighbors of the same class are penalized [4]. The algorithm samples random observations and locates their nearest neighbors from the same and opposite classes. The values of the features of the nearest neighbors are compared to the sampled instance and used to update the relevance scores for each feature. Relief-F is sensitive to features interaction without evaluating combinations of features [51], making it computationally efficient (its complexity is O(n2p), where n is the number of observations), yet sensitive to complex patterns of associations. Unlike mRMR, Relief-F does not remove redundant features, but adding features that are presumably redundant can lead to noise reduction and better class separation, as very high variable correlation (or anti-correlation) does not exclude variable complementarity [21]. Relief-F is widely used because of its simplicity, high operation efficiency and applicability to multiclass problems [52].
- Chi-squared: an individual Chi-squared test evaluates the relationship between each feature and the target classes: when they are independent, the statistics takes on a low value (high p-value), while high values indicate that the hypothesis of independence between the feature and the target class can be rejected (small p-value). Since Chi-squared test works for categorical predictors and gene expression data are continuous variables, we binned the expression values of each gene (10 bins) before each test. Unlike mRMR and Relief-F, Chi-squared is a univariate technique and evaluates each feature independently of the others.
4.2.2. Genetic Algorithms for Feature Selection
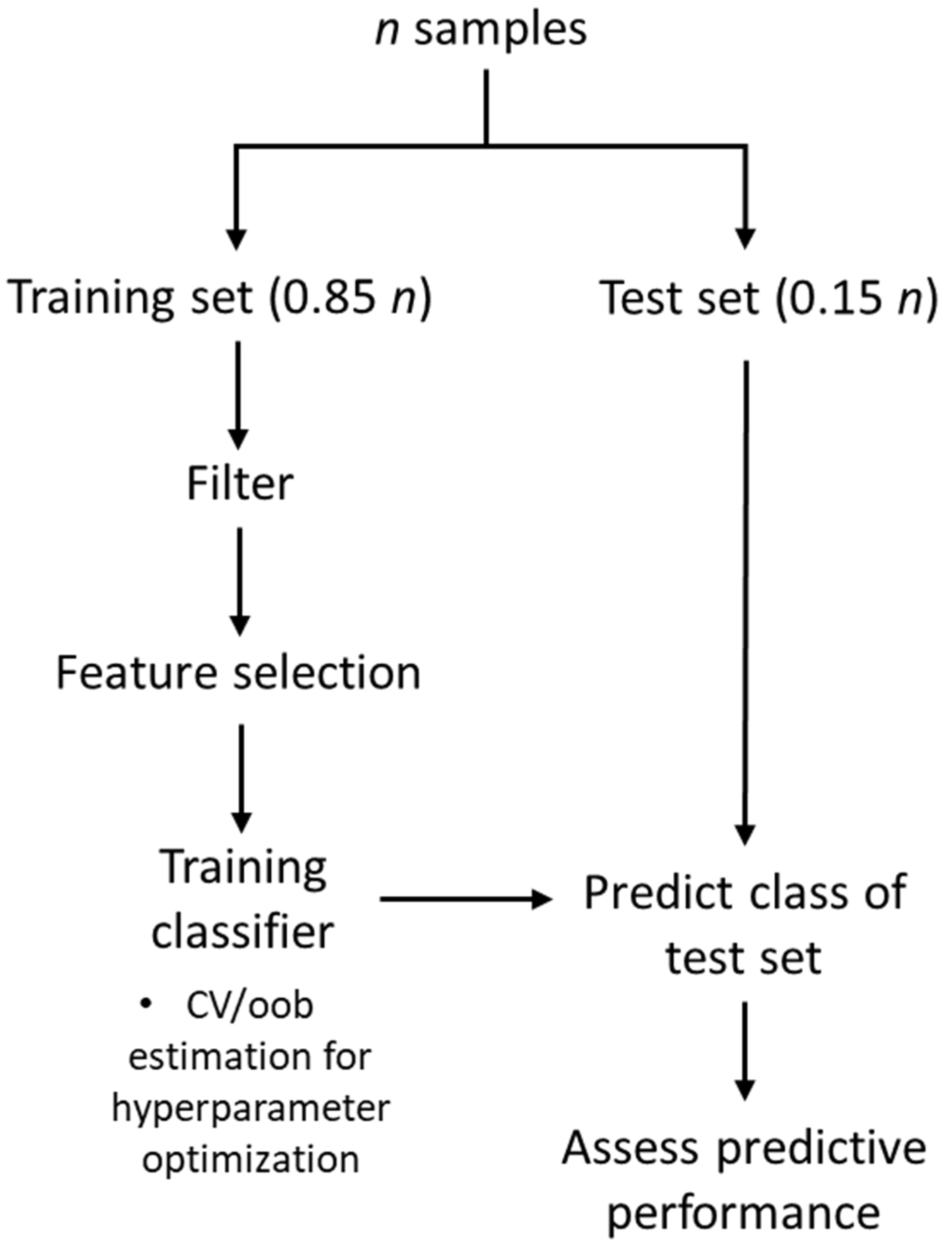
4.3. Workflow
4.4. Datasets
4.5. Performance Assessment Criteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Classification Learning Algorithms
- RF: class prediction with RF is achieved as follows: for each class and each tree t = 1, …, T we compute , the estimated posterior probability of class m given the observation x using tree t. Then, we average the class posterior probabilities, , over the set of selected trees as:where is the weight of tree t and is the indicator function. For each test observation, the predicted class is the one that yields the largest class posterior probability:
- PLS-DA: A number A of latent variables, where A is the hyperparameter to be optimized, are constructed by maximizing the covariance between the original features and the target classes. In formulae,where is the predicted output variable and is the matrix of coefficient of the PLS-DA model, computed in terms of the standardized gene expression matrix X, and the weights (W) and loadings (P and Q) matrices.
- SVM: given a set of observation-label pairs , where and as the training data, the equation of the class separating hyperplane for the linear SVMs employed in our study is the solution of the following optimization problem (in primal form):where w is the weight of each feature, b the bias, is the Box Constraint (a penalty parameter) of the error term, is a slack variable introduced to cast the problem in a soft-margin fashion to deal with non-separable data. Solution of (A5) yields the decision boundary with the largest margin between the two classes. The classification of a test sample x is given by:where the hat denotes the estimated optimal value for the parameter.
- RLR: Regularized Logistic Regression (RLR) [44] measures the relationship between a categorical dependent variable and one or more features, by estimating probabilities using a logit link function:where is the bias and are the coefficients for each feature . Here we employed a L1-regularization, more robust to irrelevant features and noise in the data [45]. We fitted the penalized maximum-likelihood coefficients by solving the following optimization problem:where is the regularization coefficient (the bias is excluded from the regularization term). The deviance is computed as , i.e., the difference between the loglikelihood of the model and a saturated one with the maximum number of parameters that can be estimated. When generalizing to the multiclass case M > 2, we employed the multinomial logistic regression conceived as M−1 independent binary logistic models:where is the vector of features for observation i, is a vector of regression coefficients corresponding to class m, and is the score associated with assigning observation i to class m. We estimated the unknown parameters in each vector by optimizing the following:
Appendix A.2. Genetic Algorithms for Feature Selection
- Initialization: each individual/set of retained features, is initialized at random.
- Fitness assignment: a value for the objective function is assigned to each individual in the population. Low classification errors correspond to high fitness and vice-versa. Individuals with greater fitness have higher probability of being selected for recombination at Step 4.
- Selection: at each iteration, a selection rule passes individuals to the next generation based on their fitness.
- Crossover: a crossover rule recombines the selected individuals (parents) to generate a new population. The recombination consists in picking and combining two individuals at random to get an offspring, until the new population has the same size as the old one.
- Mutation: while crossover generates offspring that are similar to the parent, mutation adds a source of variation, changing some of the included features in the offspring at random.
References
- Abusamra, H. A comparative study of feature selection and classification methods for gene expression data of glioma. Procedia Comput. Sci. 2013, 23, 5–14. [Google Scholar] [CrossRef]
- Rostami, M.; Forouzandeh, S.; Berahmand, K.; Soltani, M.; Shahsavari, M.; Oussalah, M. Gene selection for microarray data classification via multi-objective graph theoretic-based method. Artif. Intell. Med. 2022, 123, 102228. [Google Scholar] [CrossRef]
- Alhenawi, E.; Al-Sayyed, R.; Hudaib, A.; Mirjalili, S. Feature selection methods on gene expression microarray data for cancer classification: A systematic review. Comput. Biol. Med. 2022, 140, 105051. [Google Scholar] [CrossRef]
- Bolón-Canedo, V.; Sánchez-Maroño, N.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. A review of microarray datasets and applied feature selection methods. Inf. Sci. 2014, 282, 111–135. [Google Scholar] [CrossRef]
- Mahin, K.F.; Robiuddin, M.; Islam, M.; Ashraf, S.; Yeasmin, F.; Shatabda, S. PanClassif: Improving pan cancer classification of single cell RNA-seq gene expression data using machine learning. Genomics 2022, 114, 110264. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Uziela, K.; Honkela, A. Probe Region Expression Estimation for RNA-Seq Data for Improved Microarray Comparability. PLoS ONE 2015, 10, e0126545. [Google Scholar] [CrossRef]
- Microarray Analysis—Latest Research and News|Nature. Available online: https://www.nature.com/subjects/microarray-analysis (accessed on 4 September 2021).
- Moreno-Torres, J.G.; Raeder, T.; Alaiz-Rodríguez, R.; Chawla, N.V.; Herrera, F. A unifying view on dataset shift in classification. Pattern Recognit. 2012, 45, 521–530. [Google Scholar] [CrossRef]
- Liu, S.; Xu, C.; Zhang, Y.; Liu, J.; Yu, B.; Liu, X.; Dehmer, M. Feature selection of gene expression data for Cancer classification using double RBF-kernels. BMC Bioinform. 2018, 19, 396. [Google Scholar] [CrossRef]
- Li, Z.; Xie, W.; Liu, T. Efficient feature selection and classification for microarray data. PLoS ONE 2018, 13, e0202167. [Google Scholar] [CrossRef]
- Michiels, S.; Koscielny, S.; Hill, C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 2005, 365, 488–492. [Google Scholar] [CrossRef]
- James, A.P.; Dimitrijev, S. Nearest Neighbor Classifier Based on Nearest Feature Decisions. Comput. J. 2012, 55, 1072–1087. [Google Scholar] [CrossRef]
- James, A.; Dimitrijev, S. Inter-image outliers and their application to image classification. Pattern Recognit. 2010, 43, 4101–4112. [Google Scholar] [CrossRef]
- James, A.P.; Dimitrijev, S. Ranked selection of nearest discriminating features. Hum.-Cent. Comput. Inf. Sci. 2012, 2, 12. [Google Scholar] [CrossRef]
- Mitchell, T.M. Generalization as search. Artif. Intell. 1982, 18, 203–226. [Google Scholar] [CrossRef]
- Blum, A.L.; Langley, P. Selection of relevant features and examples in machine learning. Artif. Intell. 1997, 97, 245–271. [Google Scholar] [CrossRef]
- Kohavi, R.; John, G.H. Wrappers for feature subset selection. Artif. Intell. 1997, 97, 273–324. [Google Scholar] [CrossRef]
- Bolón-Canedo, V.; Seth, S.; Sánchez-Maroño, N.; Alonso-Betanzos, A.; Príncipe, J.C. Statistical dependence measure for feature selection in microarray datasets. In Proceedings of the 19th European Symposium on Artificial Neural Networks, Computational Intelligence and Machine Learning, ESANN 2011, Bruges, Belgium, 27–29 April 2011; pp. 23–28. [Google Scholar]
- Iguyon, I.; Elisseeff, A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar] [CrossRef][Green Version]
- Song, Q.; Ni, J.; Wang, G. A Fast Clustering-Based Feature Subset Selection Algorithm for High-Dimensional Data. IEEE Trans. Knowl. Data Eng. 2013, 25, 1–14. [Google Scholar] [CrossRef]
- Mirzaei, G.; Adeli, H. Machine learning techniques for diagnosis of alzheimer disease, mild cognitive disorder, and other types of dementia. Biomed. Signal Process. Control 2022, 72, 103293. [Google Scholar] [CrossRef]
- Kuncheva, L.I.; Matthews, C.E.; Arnaiz-González, Á.; Rodríguez, J.J. Feature Selection from High-Dimensional Data with Very Low Sample Size: A Cautionary Tale. arXiv 2020, arXiv:2008.12025. [Google Scholar]
- Spira, A.; Beane, J.E.; Shah, V.; Steiling, K.; Liu, G.; Schembri, F.; Gilman, S.; Dumas, Y.M.; Calner, P.; Sebastiani, P.; et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 2007, 13, 361–366. [Google Scholar] [CrossRef]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef]
- Haslinger, C.; Schweifer, N.; Stilgenbauer, S.; Döhner, H.; Lichter, P.; Kraut, N.; Stratowa, C.; Abseher, R. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J. Clin. Oncol. 2004, 22, 3937–3949. [Google Scholar] [CrossRef]
- Nematzadeh, H.; Enayatifar, R.; Mahmud, M.; Akbari, E. Frequency based feature selection method using whale algorithm. Genomics 2019, 111, 1946–1955. [Google Scholar] [CrossRef]
- Hall, M.A. Correlation-Based Feature Selection for Machine Learning. Ph.D. Dissertation, University of Waikato, Hamilton, New Zealand, 2003. Available online: https://www.cs.waikato.ac.nz/~mhall/thesis.pdf (accessed on 10 August 2022).
- Nosrati, V.; Rahmani, M. An ensemble framework for microarray data classification based on feature subspace partitioning. Comput. Biol. Med. 2022, 148, 105820. [Google Scholar] [CrossRef]
- Zhu, X.; Ying, C.; Wang, J.; Li, J.; Lai, X.; Wang, G. Ensemble of ML-KNN for classification algorithm recommendation. Knowl.-Based Syst. 2021, 221, 106933. [Google Scholar] [CrossRef]
- Aalaei, S.; Shahraki, H.; Rowhanimanesh, A.; Eslami, S. Feature selection using genetic algorithm for breast cancer diagnosis: Experiment on three different datasets. Iran. J. Basic Med. Sci. 2016, 19, 476–482. [Google Scholar]
- Bolón-Canedo, V.; Alonso-Betanzos, A. Ensembles for feature selection: A review and future trends. Inf. Fusion 2019, 52, 1–12. [Google Scholar] [CrossRef]
- Hall, M.; Smith, L.A. Practical Feature Subset Selection for Machine Learning. In Proceedings of the 21st Australasian Computer Science Conference, ACSC’98, Perth, Australia, 4–6 February 1998; 1998; pp. 181–191. [Google Scholar]
- Ramírez-Gallego, S.; Lastra, I.; Martínez-Rego, D.; Bolón-Canedo, V.; Benítez, J.M.; Herrera, F.; Alonso-Betanzos, A. Fast-mRMR: Fast Minimum Redundancy Maximum Relevance Algorithm for High-Dimensional Big Data. Int. J. Intell. Syst. 2017, 32, 134–152. [Google Scholar] [CrossRef]
- Rostami, M.; Forouzandeh, S.; Berahmand, K.; Soltani, M. Integration of multi-objective PSO based feature selection and node centrality for medical datasets. Genomics 2020, 112, 4370–4384. [Google Scholar] [CrossRef]
- Breiman, L. Statistical modeling: The two cultures (with comments and a rejoinder by the author). Stat. Sci. 2001, 16, 199–215. [Google Scholar] [CrossRef]
- Stąpor, K. Evaluating and Comparing Classifiers: Review, Some Recommendations and Limitations BT, Proceedings of the 10th International Conference on Computer Recognition Systems CORES 2017; Kurzynski, M., Wozniak, M., Burduk, R., Eds.; Springer: Cham, Switzerland, 2018; pp. 12–21. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Höskuldsson, A. PLS regression methods. J. Chemom. 1988, 2, 211–228. [Google Scholar] [CrossRef]
- Lee, L.C.; Liong, C.-Y.; Jemain, A.A. Partial least squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: A review of contemporary practice strategies and knowledge gaps. Analyst 2018, 143, 3526–3539. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, C.; Wang, H.; Wang, L.; Dai, L. PLS-DA and Vis-NIR spectroscopy based discrimination of abdominal tissues of female rabbits. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 271, 120887. [Google Scholar] [CrossRef]
- Salehi, F.; Abbasi, E.; Hassibi, B. The Impact of Regularization on High-Dimensional Logistic Regression. arXiv 2019, arXiv:1906.03761. [Google Scholar] [CrossRef]
- Vinga, S. Structured sparsity regularization for analyzing high-dimensional omics data. Brief. Bioinform. 2021, 22, 77–87. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Logistic regression. Nat. Methods 2016, 13, 541–542. [Google Scholar] [CrossRef]
- Xing, W.; Bei, Y. Medical Health Big Data Classification Based on KNN Classification Algorithm. IEEE Access 2020, 8, 28808–28819. [Google Scholar] [CrossRef]
- Classification Using Nearest Neighbors—MATLAB & Simulink—MathWorks Italia. Available online: https://it.mathworks.com/help/stats/classification-using-nearest-neighbors.html#bsehylk (accessed on 4 September 2021).
- Pozzoli, S.; Soliman, A.; Bahri, L.; Branca, R.M.; Girdzijauskas, S.; Brambilla, M. Domain expertise–agnostic feature selection for the analysis of breast cancer data. Artif. Intell. Med. 2020, 108, 101928. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Urbanowicz, R.J.; Meeker, M.; La Cava, W.; Olson, R.S.; Moore, J.H. Relief-based feature selection: Introduction and review. J. Biomed. Inform. 2018, 85, 189–203. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Chai, Z. A novel random multi-subspace based ReliefF for feature selection. Knowl.-Based Syst. 2022, 252, 109400. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Holland, J.H. Genetic Algorithms and Machine Learning Metaphors. Mach. Learn. 1988, 3, 95–99. [Google Scholar] [CrossRef]
- Leardi, R.; Boggia, R.; Terrile, M. Genetic algorithms as a strategy for feature selection. J. Chemom. 1992, 6, 267–281. [Google Scholar] [CrossRef]
- Genetic Algorithm Options—MATLAB & Simulink—MathWorks Italia. Available online: https://it.mathworks.com/help/gads/genetic-algorithm-options.html (accessed on 4 September 2021).
- Breiman, L. Out-of-Bag Estimation. 1996. Available online: https://www.stat.berkeley.edu/~breiman/OOBestimation.pdf (accessed on 10 August 2022).
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Ballabio, D.; Consonni, V. Classification tools in chemistry. Part 1: Linear models. PLS-DA. Anal. Methods 2013, 5, 3790–3798. [Google Scholar] [CrossRef]
- Qian, J.; Hastie, T.; Friedman, J.; Tibshirani, R.; Simon, N. Glmnet for Matlab. Available online: https://web.stanford.edu/~hastie/glmnet_matlab/ (accessed on 5 September 2021).
- Opitz, J.; Burst, S. Macro F1 and Macro F1. arXiv 2019, arXiv:1911.03347. [Google Scholar] [CrossRef]







| Accuracy | Recall | Precision | Specificity | NPV | F1 Score | ||
|---|---|---|---|---|---|---|---|
| RF | 67.93 ± 1.58 | 61.24 ± 2.58 | 67.6 ± 2.01 | 73.72 ± 2.09 | 69.08 ± 1.62 | 63.55 ± 1.99 | |
| PLS-DA | 92.97 ± 1.05 | 93.53 ± 2.02 | 92.97 ± 1.73 | 92.43 ± 2.02 | 95.29 ± 1.4 | 92.46 ± 1.15 | |
| SMK_CAN_187 | SVM | 68.41 ± 1.61 | 60.14 ± 2.56 | 69.37 ± 2.32 | 75.6 ± 2.41 | 68.9 ± 1.51 | 63.52 ± 2.04 |
| RLR | 68.38 ± 1.59 | 65.73 ± 2.56 | 66.83 ± 2.01 | 70.63 ± 2.26 | 70.96 ± 1.78 | 65.59 ± 1.85 | |
| kNN | 68.9 ± 1.57 | 60.07 ± 2.31 | 70.03 ± 2.17 | 76.55 ± 2.17 | 69.04 ± 1.49 | 64 ± 1.85 | |
| RF | 89.85 ± 1.51 | 74.5 ± 4.37 | 87.3 ± 3.26 | 95.48 ± 1.14 | 91.54 ± 1.45 | 78.36 ± 3.42 | |
| PLS-DA | 88.85 ± 1.73 | 100 ± 0 | 73.92 ± 3.31 | 84.81 ± 2.31 | 100 ± 0 | 83.92 ± 2.23 | |
| GLI_85 | SVM | 88.77 ± 1.58 | 73.58 ± 4.34 | 84.82 ± 3.66 | 94.46 ± 1.35 | 91.04 ± 1.43 | 76.71 ± 3.48 |
| RLR | 87.15 ± 1.63 | 73.92 ± 4.64 | 79.19 ± 3.78 | 91.94 ± 1.53 | 91.18 ± 1.47 | 74.7 ± 3.44 | |
| kNN | 89.31 ± 1.63 | 70.83 ± 4.63 | 88.42 ± 3.47 | 96.2 ± 1.13 | 90.29 ± 1.51 | 76.6 ± 3.74 | |
| RF | 71.18 ± 1.78 | 78.32 ± 1.38 | 78.45 ± 1.52 | 82.7 ± 1.09 | 83.35 ± 1.09 | 77.36 ± 1.43 | |
| PLS-DA | 71.29 ± 1.97 | 78.71 ± 1.46 | 75.58 ± 2.11 | 83.3 ± 1.13 | 83.65 ± 1.13 | 75.64 ± 1.87 | |
| CLL_SUB_111 | SVM | 72.41 ± 1.89 | 79.4 ± 1.45 | 80.28 ± 1.55 | 83.35 ± 1.14 | 84.1 ± 1.14 | 78.91 ± 1.5 |
| RMR | 72.59 ± 1.85 | 78.59 ± 1.57 | 80.6 ± 1.54 | 83.49 ± 1.12 | 84.41 ± 1.12 | 78.3 ± 1.54 | |
| kNN | 69.06 ± 2.02 | 77.01 ± 1.5 | 77.34 ± 1.71 | 81.35 ± 1.22 | 82.09 ± 1.22 | 76.2 ± 1.59 |
| (A) Feature Selection | mRMR | Relief-F | Chi-Squared | Variance | |
| runtime | 00:03:23.420 | 00:00:08.355 | 00:00:00.074 | 00:00:00.038 | |
| (B) Classifier Training | RF | PLS-DA | SVM | RLR | kNN |
| runtime | 00:00:10.517 | 00:00:00.322 | 00:00:02.041 | 00:02:41.384 | 00:00:01.518 |
| (C) GA-based wrapper 50—Pop Size:50, Max Gen: 35 | RF | PLS-DA | SVM | RLR | kNN |
| runtime | 00:44:49.874 | 00:00:51.536 | 01:10:54.360 | 00:42:12.297 | 00:05:06.008 |
| (D) GA-based wrapper 50—Pop Size:200, Max Gen: 150 | RF | PLS-DA | SVM | RLR | kNN |
| runtime | 02:06:01.370 | 00:01:26.498 | 13:06:10.923 | 10:11:14.675 | 00:13:46.126 |
| Option | Setting |
|---|---|
| Population size | 50–200 |
| Max Generations | 35–150 |
| Mutation Function | ‘mutation adapt feasible’ |
| Crossover Function | crossover scattered |
| Crossover Fraction | 8% |
| Selection Function | ‘selectionstochunif’ [55] |
| Elite count | 5% of Population Size |
| RF | PLS-DA | SVM | RLR/RMR | kNN | |
|---|---|---|---|---|---|
| Hyperparameter | T | A | C | λ | k |
| Range | 1–256 | 1–15 | 10−5–103 | 1–15 | |
| Objective function | out-of-bag error | cv-error | cv-error | deviance | cv-error |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanella, L.; Facco, P.; Bezzo, F.; Cimetta, E. Feature Selection and Molecular Classification of Cancer Phenotypes: A Comparative Study. Int. J. Mol. Sci. 2022, 23, 9087. https://doi.org/10.3390/ijms23169087
Zanella L, Facco P, Bezzo F, Cimetta E. Feature Selection and Molecular Classification of Cancer Phenotypes: A Comparative Study. International Journal of Molecular Sciences. 2022; 23(16):9087. https://doi.org/10.3390/ijms23169087
Chicago/Turabian StyleZanella, Luca, Pierantonio Facco, Fabrizio Bezzo, and Elisa Cimetta. 2022. "Feature Selection and Molecular Classification of Cancer Phenotypes: A Comparative Study" International Journal of Molecular Sciences 23, no. 16: 9087. https://doi.org/10.3390/ijms23169087
APA StyleZanella, L., Facco, P., Bezzo, F., & Cimetta, E. (2022). Feature Selection and Molecular Classification of Cancer Phenotypes: A Comparative Study. International Journal of Molecular Sciences, 23(16), 9087. https://doi.org/10.3390/ijms23169087