Abstract
The inositol 1,4,5-triphosphate receptor type 1 (ITPR1) gene encodes an InsP3-gated calcium channel that modulates intracellular Ca2+ release and is particularly expressed in cerebellar Purkinje cells. Pathogenic variants in the ITPR1 gene are associated with different types of autosomal dominant spinocerebellar ataxia: SCA15 (adult onset), SCA29 (early-onset), and Gillespie syndrome. Cerebellar atrophy/hypoplasia is invariably detected, but a recognizable neuroradiological pattern has not been identified yet. With the aim of describing ITPR1-related neuroimaging findings, the brain MRI of 14 patients with ITPR1 variants (11 SCA29, 1 SCA15, and 2 Gillespie) were reviewed by expert neuroradiologists. To further evaluate the role of superior vermian and hemispheric cerebellar atrophy as a clue for the diagnosis of ITPR1-related conditions, the ITPR1 gene was sequenced in 5 patients with similar MRI pattern, detecting pathogenic variants in 4 of them. Considering the whole cohort, a distinctive neuroradiological pattern consisting in superior vermian and hemispheric cerebellar atrophy was identified in 83% patients with causative ITPR1 variants, suggesting this MRI finding could represent a hallmark for ITPR1-related disorders.
1. Introduction
The inositol 1,4,5-triphosphate receptor type 1 (ITPR1) gene encodes a ligand-gated calcium channel localized in the endoplasmic reticulum membrane, which modulates intracellular Ca2+ release, representing a signaling hub for the cell [1,2]. ITPR1 is widely expressed in the brain, with highest expression in cerebellar Purkinje cells. Heterozygous pathogenic variants of ITPR1 have been associated with a broad clinical spectrum, ranging from adult-onset Spinocerebellar Ataxia type 15 (SCA15) to early-onset Spinocerebellar Ataxia type 29 (SCA29) and Gillespie syndrome, while homozygous variants have been described only in the early onset form [3]. SCA29 is characterized by onset in infancy or childhood of ataxia and other cerebellar signs such as hypotonia and oculomotor abnormalities, frequently associated with delayed motor development and cognitive impairment [1]. Gillespie syndrome is characterized by a more severe phenotype featuring the cerebellar syndrome, intellectual disability, and aniridia [3]. Both familial and sporadic cases with early-onset cerebellar ataxia associated with ITPR1 gene mutations have been reported to date [4]. According to available studies, brain magnetic resonance imaging (MRI) of ITPR1-related disorders is characterized by cerebellar atrophy as the main finding. Extra-cerebellar findings have been rarely reported, involving both supratentorial regions and pons [4,5,6,7,8]. Most of the studies describe a pattern of cerebellar atrophy which is more severe in the vermis than in the cerebellar hemispheres, while only one patient with a pattern consistent with Ponto Cerebellar Hypoplasia (i.e., “dragonfly” cerebellum) has been reported so far [8]. In some cases, a progression of cerebellar atrophy over time is observed [2,4,6,7,9]. More recently, the pattern of atrophy was noted to be more severe in the superior part of the hemispheres (and vermis) than in the inferior part [4,6,9]. According to these observations, we reviewed a cohort of ITPR1-mutated patients to better define the characteristics of cerebellar alterations. In addition, genetic analysis of the ITPR1 gene was performed on patients with superior cerebellar atrophy at MRI to establish the value of this sign as a predictive clue for the diagnosis of ITPR1 gene-related disorders.
2. Materials and Methods
Three different Italian centers participated in this retrospective study: Scientific Institute E. Medea, Bosisio Parini (LC), Bambino Gesù Children’s Hospital, and Romeand Neurological Scientific Institute C. Mondino, Pavia. Two different groups of subjects were recruited. Group A included patients already diagnosed with a pathogenetic ITPR1 gene variant through a clinical diagnostic test, while Group B included subjects without a genetic diagnosis, who presented a brain MRI pattern of isolated superior hemispheric and vermian cerebellar atrophy, identified through an extensive review of our neuroradiological databases. All the available images of Group A patients were collected and reviewed independently by two experts in pediatric neuroradiology to assess the cerebellar and cerebral findings. In particular, sagittal and coronal slices were used to evaluate the presence, distribution, and severity of cerebellar atrophy in the upper and lower cerebellum. Signal alterations of cerebellar cortex and white matter as well as supratentorial findings were also investigated. When multiple studies were available, the progression of atrophy was recorded. Patients of Group B were first identified by querying the neuroimaging databases, selecting those patients affected by predominantly or exclusively superior cerebellar atrophy with no history of acquired conditions (e.g., infective, ischemic etc.). Images were then reviewed to confirm the pattern of cerebellar atrophy predominantly involving the superior part of the hemisphere and vermis and selected subjects underwent genetic analysis of the ITPR1 gene. Only selected patients for whom DNA material was available were finally included in Group B.
For genetic studies, genomic DNA was extracted from peripheral blood of the patients and their parents. Next Generation Sequencing (NGS) analysis was performed either on targeted panels of genes causative of various forms of cerebellar ataxias (16 patients), clinical exome (1 patient), or whole exome (2 patients). The targeted gene panels were sequenced using either Nextera (Illumina, San Diego, CA, USA) or SureSelect (Agilent Technologies, Santa Clara, CA, USA) enrichment protocols and run on MiSeq or NextSeq sequencing platforms (Illumina, San Diego, CA, USA), with an expected coverage of >99% of targeted genomic regions. For clinical exome and whole exome, DNA libraries were amplified using the SureSelectXT Focused Exome (Agilent Technologies) and Twist Human Core Kit (Twist Bioscience, South San Francisco, CA, USA), respectively, and sequenced on a NextSeq platform (Illumina). Bioinformatic analysis was carried out by aligning sequences to the human reference genome (GRCh37) using Bowtie2 or BWA v0.7.5. ANNOVAR and GATK Unified Genotyper were used to call variants, which were annotated through the eVANT v1.3 software (enGenome, Pavia, Italy). Subsequent filtering steps allowed to exclude intronic variants, synonymous variants not affecting splicing, and variants with frequency > 1% in human variation databases. We used several in silico tools to predict pathogenicity of identified variants, including Deleterious Annotation of genetic variants using Neural Networks (DANN), Combined Annotation-Dependent Depletion (CADD), Polymorphism Phenotyping v2 (PolyPhen-2), and Sorting Intolerant from Tolerant (SIFT). Variants were classified according to the American College of Human Society (ACMG) guidelines. Segregation was verified by Sanger sequencing in the families. Accession numbers are the following: human ITPR1 mRNA: NM_001168272.1; human ITPR1 protein: NP_001161744.1.
3. Results
3.1. Demographic Data
Group A included 14 patients (8 females and 6 males) from 10 unrelated families, with average age at the last follow-up of 18 years (min 2 years; max 56 years). One patient received a diagnosis of SCA15 (patient 4), eleven had SCA29, while two (patient 6 and 10) had Gillespie syndrome.
Six patients (5 females and 1 male, all sporadic) with a superior cerebellar atrophy were initially identified but due to lack of DNA from one patient, Group B finally included five patients. The average age at last follow-up was 7 years (min 6 months; max 14 years).
All patients belonging to Group B had a SCA29 phenotype (see Table 1).

Table 1.
Clinical, neuroradiological and genetic features of enrolled patients.
3.2. Genetic Data
Pathogenic or likely pathogenic variants in the ITPR1 gene were overall detected in 16 patients, while 2 siblings carried a novel variant of unknown significance (VUS) (p.S695N; CADD 20.7), which was absent from our in-house database of over 2000 WES, as well as from the gnomAD population database (Figure 1, Table 1).
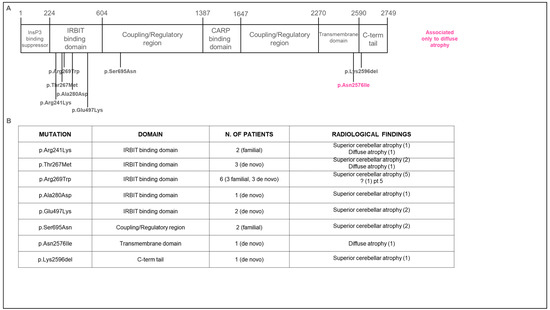
Figure 1.
Genetic findings in enrolled patients. (A) Schematic representation of the position of variants within functional domains of the ITPR1 protein. Most variants are located in the IRBIT domain, suggesting that loss of the channel function impairs the IP3-induced Ca2+ release. (B) Summary of variants and radiological features found in our cohort.
In Group A, only the p.S695N missense variant was novel, while all other missense variants (p.R269W, p.T267M, p.R241K, p.N2576I, p.A280D, p.E497K) as well as a one-amino acid deletion (p.K2596del) had already been reported [1,9,10,11].
In Group B, 5 patients were genetically tested, of whom 4 were found to carry the following ITPR1 previously reported missense variants: p.R269W; p.T267M (2 unrelated patients); p.E497K [1,10].
3.3. Neuroradiological Data
In Group A, 11/14 patients showed a pattern of predominant superior cerebellar atrophy (very mild to severe) while 3/14 patients showed diffuse atrophy.
In cases of superior atrophy, both the upper part of the vermis and hemispheres were affected (Figure 2). Follow-up studies were available in three cases: in one case, diffuse cerebellar atrophy became evident between 5 months and 6 years of age (Figure 3A); in the second case, superior atrophy was not present at 8 months of age (Figure 3B) and it became evident at 3 years of age; in the last one, very mild superior cerebellar atrophy remained stable over a 4 year period (Figure 3D). Regarding signal alterations, a very mild hyperintensity of the superior cerebellar cortex close to the vermis was noticed in three cases on FLAIR but not on T2-weighted images. The significance of this finding remains unclear as it could be partially related to artifacts at the interface between cortex and enlarged CSF spaces. Supratentorial findings were mostly normal; in two patients from the same family, a dysmorphic corpus callosum with enlarged lateral ventricles was observed.
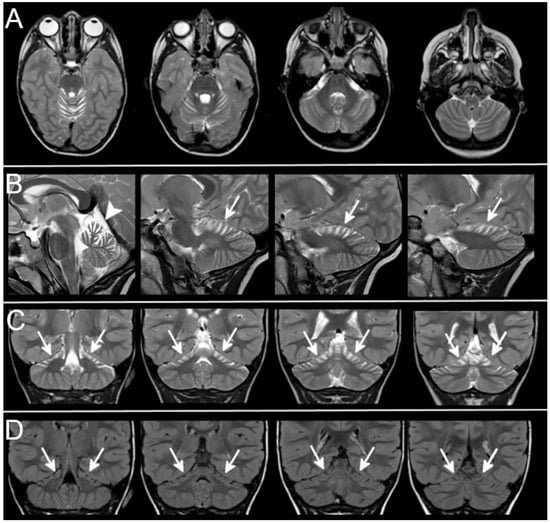
Figure 2.
Superior cerebellar atrophy. Axial (A), sagittal (B), coronal (C), T2-weighted and coronal FLAIR (D) sections show the typical pattern of superior cerebellar atrophy (Patient 18-Table 1). The superior part of cerebellar hemispheres (arrows) and vermis (arrowhead) show marked atrophy with enlarged cortical CSF spaces. No cerebellar signal alterations can be detected on T2-weighted and FLAIR sections. The inferior part of the cerebellum is not atrophic and looks normal.
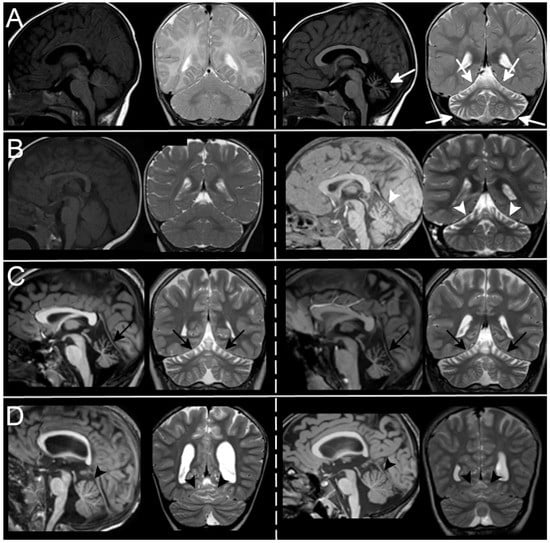
Figure 3.
MRI evolution over time. In (A), Patient7-Table 1 has a normal cerebellum at 5 months of age (left) while he shows a diffuse cerebellar atrophy (white arrows) at 6 years of age (atrophy was also present at 2 years of age. Not shown here). In (B), Patient 12-Table 1 develops superior cerebellar atrophy (white arrowheads) between 8 months of age (left, normal cerebellum), and 3 years (right, cerebellar atrophy). No progression of atrophy is seen in Patients 1 and 18-Table 1, with moderate ((C), black arrows) and very mild ((D), black arrowheads) superior cerebellar atrophy within a 2-year and 4-year period respectively.
Patients of Group B were retrospectively selected according to MRI reports and images. They showed in 4/5 cases a clear pattern of superior cerebellar atrophy with almost normal inferior cerebellum, and in 1/5 a diffuse atrophy, more severe in the upper cerebellum (Figure 4). One patient had a follow-up scan that did not document any progression over a 2-year period between 12 and 14 years of age (Figure 3C).
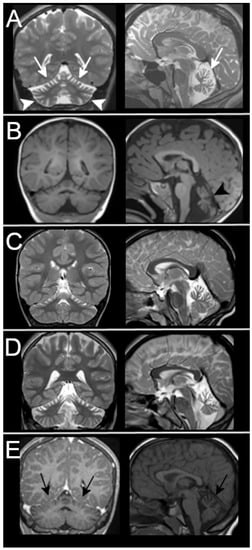
Figure 4.
MRI findings in patients from Group B. MRIs of the 5 patients retrospectively selected according to the imaging pattern are shown here. All of them have superior cerebellar atrophy. Patient 14-Table 1 in (A) shows diffuse cerebellar atrophy that also involves the inferior cerebellum (arrowheads) but that is more severe in the upper part of vermis and hemispheres (white arrows). The patient with mild superior atrophy (black arrows) in (E) tested negative for ITPR1 gene defects.
Supratentorial findings were unremarkable in all cases.
Neuroradiological, genetic, and clinical data of all patients with ITPR1 variants reported in the literature are summarized in Table 2.
Table 2.
Gene mutations described up to date.
4. Discussion
Imaging findings in ITPR1-mutated patients are sparsely reported in the literature, and not systematically addressed [8]. In the available studies, the most frequently described neuroradiological feature is a diffuse cerebellar atrophy, sometimes reported as predominantly involving the vermis, and just in a minority of cases involving the superior vermis and hemispheres [3,9,10,11,15].
In our study population, all three ITPR1-related phenotypes were represented. We included both patients previously diagnosed with a pathogenic or likely pathogenic ITPR1 variant as well as patients who were directed to ITPR1 genetic testing due to the presence of isolated superior vermian and hemispheric cerebellar atrophy. We also included two affected siblings carrying a novel ITPR1 variant classified as VUS (p.S695N), since the clinical and neuroradiological phenotype in both patients were highly suggestive of an ITPR1-related defect. Unfortunately, the family is from Morocco and parents were not available for clinical examination and segregation analysis.
Considering all ITPR1 mutated patients, a characteristic pattern of superior vermian and cerebellar atrophy was present in 83%, while in the remaining cases (3/18, 17%) a less peculiar diffuse cerebellar atrophy was noted. We searched for specific clinical features or a different severity manifestation in patients with diffuse cerebellar atrophy, but we could not identify differences compared to patients who showed the more typical superior cerebellar involvement [14,16,17,18,19].
ITPR1 gene encodes for inositol 1,4,5-triphosphate receptor type 1, an InsP3-gated calcium channel that modulates intracellular Ca2+ release and plays the crucial role in the regulation of spine distribution and morphology of adult Purkinje cells [20]. The primary structure of the protein ITPR1 consists of three domains, including an InsP3-binding domain in the N-terminus, a regulatory carbonic anhydrase-related protein VIII (CARP)-binding domain, and a transmembrane-spanning domain near the C-terminus [1]. According to the literature, most of the pathogenic variants in our cohort (as the majority of reported variants) fall within the IRBIT binding domain, where lie most of the mutations associated with SCA29, while variants that cause Gillespie syndrome are mostly located at the C-terminus of the protein, especially in the transmembrane domain [9]. In our cohort, we observed intrafamiliar variability both in terms of clinical phenotype and neuroradiological pattern. For instance, in family V, the proband (patient 8) showed a phenotype compatible with SCA29, while the mother (patient 9) had a later-onset, milder phenotype with only slurred speech, suggesting a SCA15 phenotype. This evidence highlights the possible occurrence of distinct phenotypes (SCA15 and SCA29) in association with the same mutation, as previously suggested [1].
Of note, we observed a variable expression of the neuroradiological pattern among patients carrying the same ITPR1 variant, with the only exception of carriers of the recurrent p.R269W variant, who showed a fully concordant imaging phenotype of superior cerebellar atrophy. Overall, we failed to detect reliable correlates between the protein domain harboring the mutation and the pattern of cerebellar atrophy observed (diffused vs. predominantly superior). We speculate that other regulatory factors might influence the pattern of expression of the protein in the cerebellum, as already suggested by Kerkhofs et al. [21].
Aside from the characterization of neuroimaging pattern, the analysis of Group B patients highlights the importance of recognizing superior cerebellar atrophy as a diagnostic clue for ITPR1-related disorders, since four out of five subjects presenting this peculiar imaging trait, retrospectively selected from two large imaging databases, tested positive for pathogenic variants in the ITPR1 gene. Predominant atrophy of the upper parts of the cerebellum has never been previously associated to proven genetic conditions [22,23], while superior vermian atrophy has been described in neonates suffering from hypoxic-ischemia [24]. This represents a novel and key element for improving the diagnosis of children with either static or progressive cerebellar atrophy.
Clinical features of pediatric ataxia are usually unspecific and can be ascribed to a very large number of genetic conditions. Generally, the initial diagnostic workup, beyond a comprehensive clinical assessment of ataxic and non ataxic symptoms, includes neuroimaging and genetic investigations [25]. Therefore, considering our finding, an MRI pattern of mild to severe atrophy involving the superior part of cerebellar hemispheres and vermis (typically without any signal alterations within the cortex), with normal supratentorial brain and without history and cerebral signs of hypoxic-ischemic injury, might represent a very important insight and should prompt suspicion of a possible ITPR1 gene-related disorder and genetic testing is highly recommended.
The retrospective nature of this study represents a limitation for an even more extensive definition of the imaging spectrum of ITPR1-mutated patients. For instance, due to the lack of seriate MRIs in all patients, we could not establish the presence and severity of atrophy at symptoms’ onset, nor we could assess its progression over time. The few cases with follow-up imaging suggest that atrophy is not evident in the very first months of life, but it becomes evident during early childhood and may remain stable afterward. However, further confirmation is needed for this preliminary observation.
In conclusion, through a careful review of MRI images, we demonstrated a peculiar pattern of cerebellar atrophy in patients with ITPR1 gene defects and we propose that it is highly suggestive for the diagnosis, which might orient the choice of genetic testing.
Author Contributions
R.R. was responsible for data acquisition, funding, and manuscript drafting. L.P. was involved in drafting the manuscript. E.P. performed molecular study and was involved in drafting the manuscript. F.D. and V.S. performed molecular study and was involved in drafting the manuscript. C.A. was involved in data acquisition and in drafting the manuscript. S.S. was involved in data acquisition and in drafting the manuscript. L.T. was involved in data acquisition and in drafting the manuscript. E.B. was involved in critically revising the manuscript and in acquisition of funding. M.T.B. was involved in critically revising the manuscript and acquisition of funding. E.M.V. was involved in critically revising the manuscript and in acquisition of funding. G.Z. was involved in data acquisition, critically revising the manuscript, and in acquisition of funding. R.B. was involved in critically revising the manuscript and in acquisition of funding. F.A. performed the neuroradiological study and was involved in critically revising the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by funds from the Italian Ministry of Health (grant # RC2021 265 and RC2022 to IRCCS Medea and IRCCS Mondino, Ricerca Finalizzata grant number RF-2019-266 12369368 to E.M.V.), 5X MILLE (to R.R.), Fondazione Mariani (Neuropediatric Network project to E.M.V. 267 and R.B.) and funds from the Fondazione Regionale Lombarda per la Ricerca Biomedica FRRB (grant 268 # Care4NeuroRare to M.T.B.). E.B. and G.Z. are members of the European Reference Network for Rare 269 Neurological Diseases—Project ID No 739510.
Institutional Review Board Statement
The study was approved by Ethic Committe Mondino foundation (Neuropediatric Network project) number P-20200051830 All patients or their caregivers gave written informed consent for the study.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barresi, S.; Niceta, M.; Alfieri, P.; Brankovich, V.; Piccini, G.; Bruselles, A.; Barone, M.; Cusmai, R.; Tartaglia, M.; Bertini, E.; et al. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin. Genet. 2017, 91, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Warman-Chardon, J.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 2012, 7, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, J.; Ali, Z.; Farooq, M.; Khan, K.; Wikström, J.; Iqbal, M.; Zulfiqar, S.; Faryal, S.; Baig, S.M.; Dahl, N. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur. J. Hum. Genet. 2017, 25, 848–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Ohba, C.; Iai, M.; Hirabayashi, S.; Osaka, H.; Hiraide, T.; Saitsu, H.; Matsumoto, N. Sporadic infantile-onset spinocerebellar ataxia caused by missense mutations of the inositol 1,4,5-triphosphate receptor type 1 gene. J. Neurol. 2015, 262, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.J.; Sweeney, M.G.; Li, A.; Treacy, C.; Chandrashekar, H.S.; Giunti, P.; Goold, R.G.; Davis, M.B.; Houlden, H.; Tabrizi, S.J. An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: A genetic, clinical and radiological description. Mov. Disord. 2010, 25, 2176–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambonin, J.L.; Bellomo, A.; Ben-Pazi, H.; Everman, D.B.; Frazer, L.M.; Geraghty, M.T.; Harper, A.D.; Jones, J.R.; Kamien, B.; Kernohan, K.; et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: A case series and review of this emerging congenital ataxia. Orphanet J. Rare Dis. 2017, 12, 121. [Google Scholar] [CrossRef]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Brémond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, T.; Barth, P.; Reneman, L.; Appelhof, B.; Baas, F.; Poll-The, B.T. A de novo missense mutation in the inositol 1,4,5-triphosphate receptor type 1 gene causing severe pontine and cerebellar hypoplasia: Expanding the phenotype of ITPR1-related spinocerebellar ataxia’s. Am. J. Med. Genet. A 2017, 173, 207–212. [Google Scholar] [CrossRef]
- McEntagart, M.; Williamson, K.A.; Rainger, J.K.; Wheeler, A.; Seawright, A.; De Baere, E.; Verdin, H.; Bergendahl, L.T.; Quigley, A.; Rainger, J.; et al. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am. J. Hum. Genet. 2016, 98, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Ohba, C.; Osaka, H.; Iai, M.; Yamashita, S.; Suzuki, Y.; Aida, N.; Shimozawa, N.; Takamura, A.; Doi, H.; Tomita-Katsumoto, A.; et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013, 14, 225–232. [Google Scholar] [CrossRef]
- Dentici, M.L.; Barresi, S.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Bruselles, A.; Pantaleoni, F.; Danieli, A.; Iarossi, G.; Cappa, M.; et al. Identification of novel and hotspot mutations in the channel domain of ITPR1 in two patients with Gillespie syndrome. Gene 2017, 628, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 2008, 71, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Helbig, K.L.; Harmuth, F.; Deconinck, T.; Tanpaiboon, P.; Sun, B.; Guo, W.; Wang, R.; Palmaer, E.; Tang, S.; et al. De novo ITPR1 variants are a recurrent cause of early-onset ataxia, acting via loss of channel function. Eur. J. Hum. Genet. 2018, 26, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadrina, M.I.; Shulskaya, M.V.; Klyushnikov, S.; Nikopensius, T.; Nelis, M.; Kivistik, P.A.; Komar, A.A.; Limborska, S.A.; Illarioshkin, S.N.; A Slominsky, P. ITPR1 gene p.Val1553Met mutation in Russian family with mild Spinocerebellar ataxia. Cerebellum Ataxias 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, E.; Orsi, L.; Godani, M.; Vaula, G.; Jensen, S.; Salmon, E.; Ferrari, G.; Squadrone, S.; Abete, M.C.; Cagnoli, C.; et al. Two Italian Families with ITPR1 Gene Deletion Presenting a Broader Phenotype of SCA15. Cerebellum 2010, 9, 115–123. [Google Scholar] [CrossRef]
- Paganini, L.; Pesenti, C.; Milani, D.; Fontana, L.; Motta, S.; Sirchia, S.M.; Scuvera, G.; Marchisio, P.; Esposito, S.; Cinnante, C.M.; et al. A novel splice site variant in ITPR1 gene underlying recessive Gillespie syndrome. Am. J. Med. Genet. 2018, 176, 1427–1431. [Google Scholar] [CrossRef]
- Wang, L.; Hao, Y.; Yu, P.; Cao, Z.; Zhang, J.; Zhang, X.; Chen, Y.; Zhang, H.; Gu, W. Identification of a Splicing Mutation in ITPR1 via WES in a Chinese Early-Onset Spinocerebellar Ataxia Family. Cerebellum 2018, 17, 294–299. [Google Scholar] [CrossRef] [Green Version]
- Stendel, C.; Wagner, M.; Rudolph, G.; Klopstock, T. Gillespie’s Syndrome with Minor Cerebellar Involvement and No Intellectual Disability Associated with a Novel ITPR1 Mutation: Report of a Case and Literature Review. Neuropediatrics 2019, 50, 382–386. [Google Scholar] [CrossRef]
- Tada, M.; Nishizawa, M.; Onodera, O. Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias. Neurochem. Int. 2016, 94, 1–8. [Google Scholar] [CrossRef]
- Sugawara, T.; Hisatsune, C.; Le, T.D.; Hashikawa, T.; Hirono, M.; Hattori, M.; Nagao, S.; Mikoshiba, K. Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintain-ing the spine morphology of purkinje cells in adult mice. J. Neurosci. 2013, 33, 12186–12196. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, M.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G.; Parys, J.B. Pathophysiological consequences of isoform-specific IP3 receptor mutations. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865 Pt B, 1707–1717. [Google Scholar] [CrossRef]
- Poretti, A.; Wolf, N.; Boltshauser, E. Differential diagnosis of cerebellar atrophy in childhood. Eur. J. Paediatr. Neurol. 2008, 12, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Boltshauser, E.; Poretti, A. Differential Diagnosis of Cerebellar Atrophy in Childhood: An Update. Neuropediatrics 2015, 46, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M.A.; Poskitt, K.J.; Roland, E.H.; Hill, A.; Hendson, G. Cerebellar vermian atrophy after neonatal hypoxic-ischemic en-cephalopathy. AJNR Am. J. Neuroradiol. 2004, 25, 1008–1015. [Google Scholar] [PubMed]
- Brandsma, R.; Verschuuren-Bemelmans, C.; Amrom, D.; Barisic, N.; Baxter, P.; Bertini, E.; Blumkin, L.; Brankovic-Sreckovic, V.; Brouwer, O.; Bürk, K.; et al. A clinical diagnostic algorithm for early onset cerebellar ataxia. Eur. J. Paediatr. Neurol. 2019, 23, 692–706. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).