
Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders


 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Demographic Data
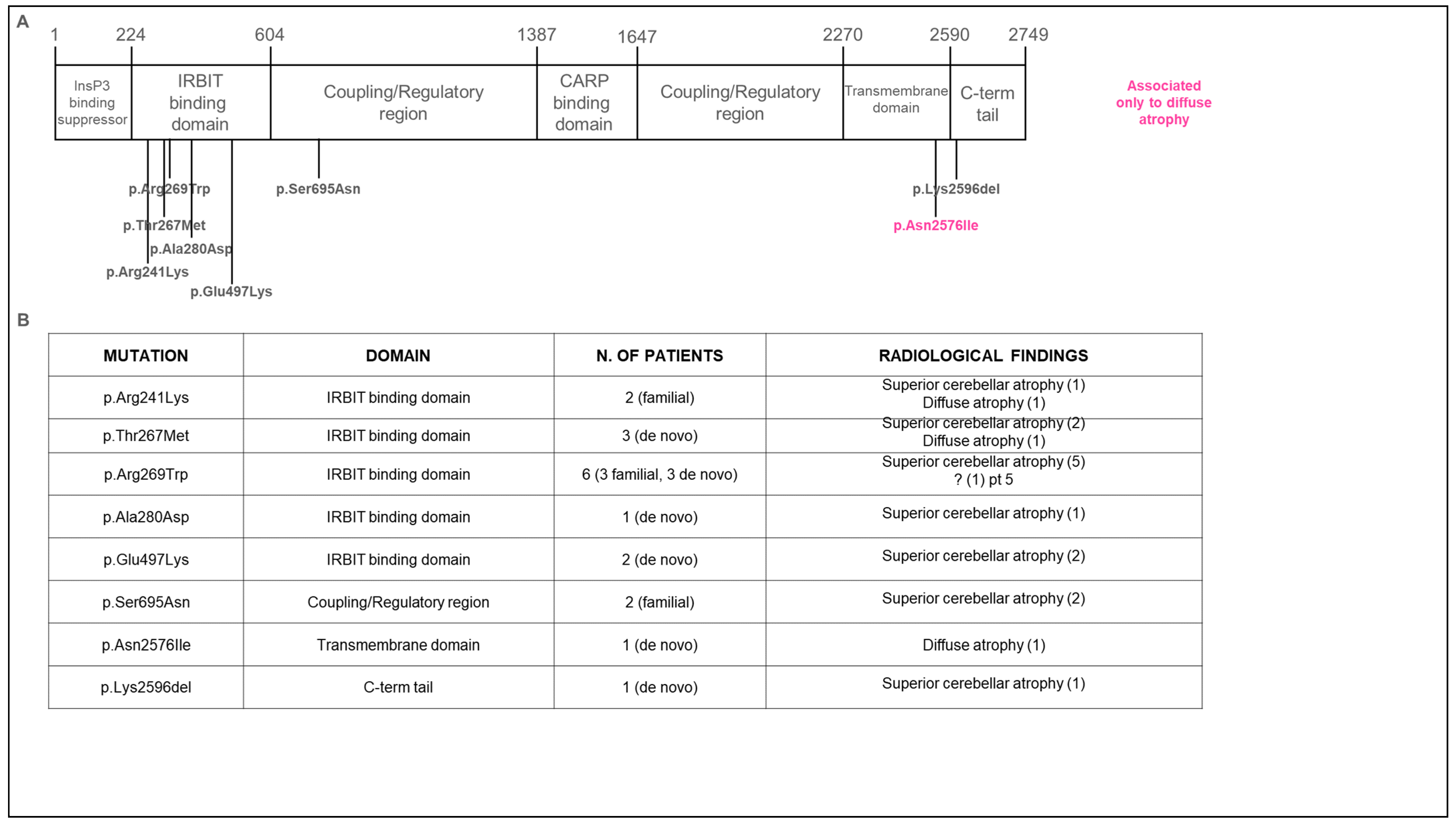
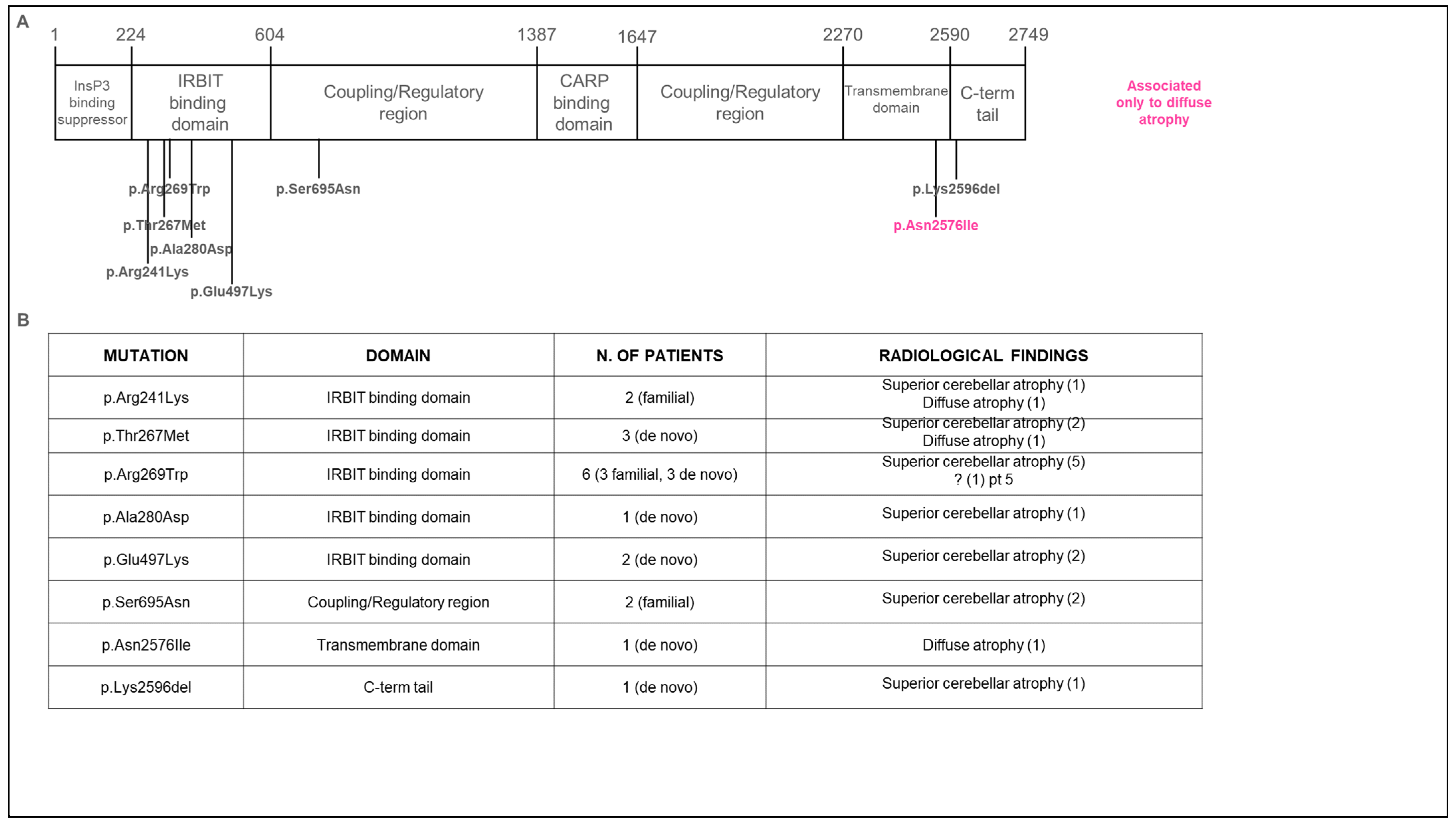
3.2. Genetic Data
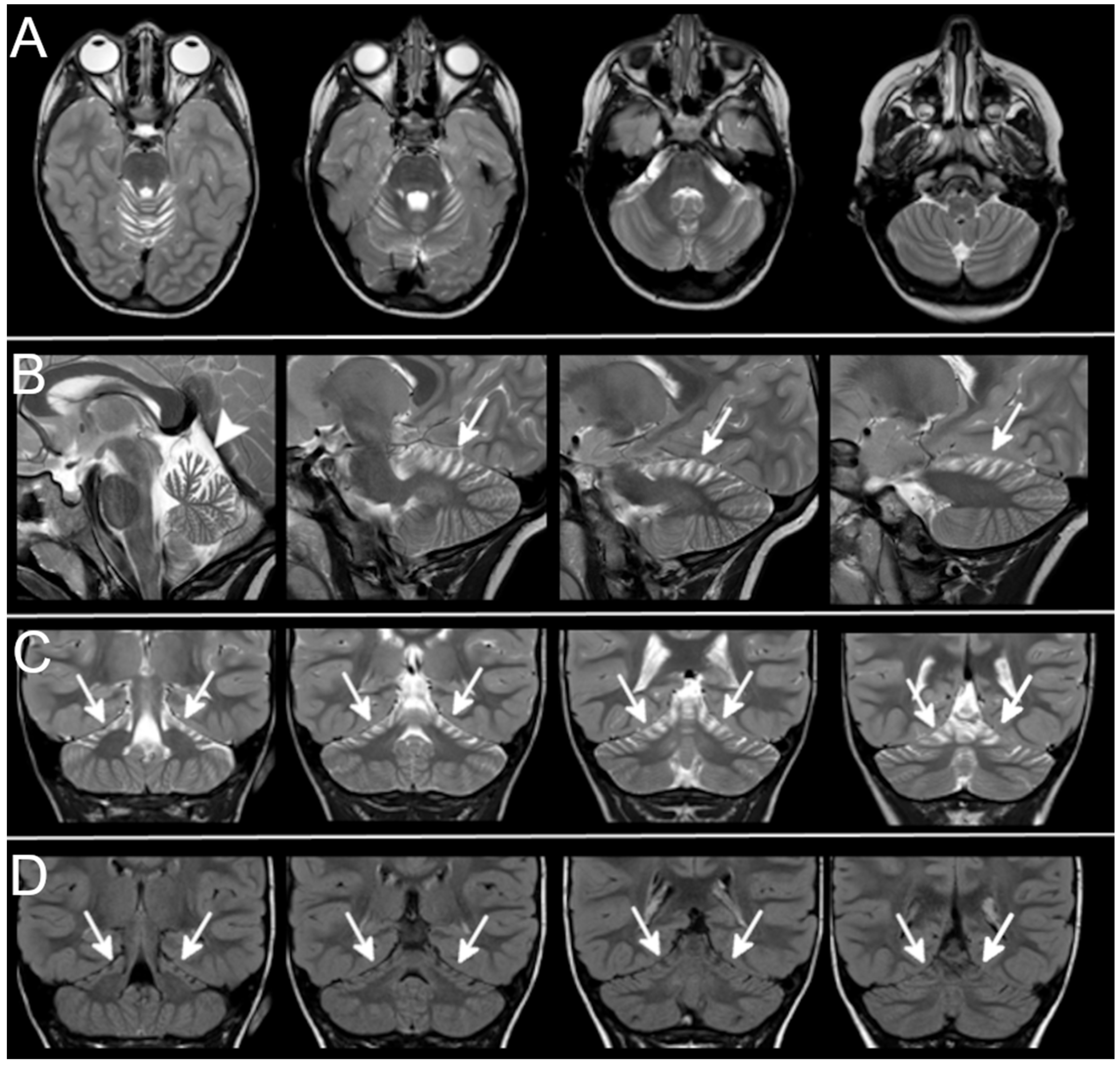
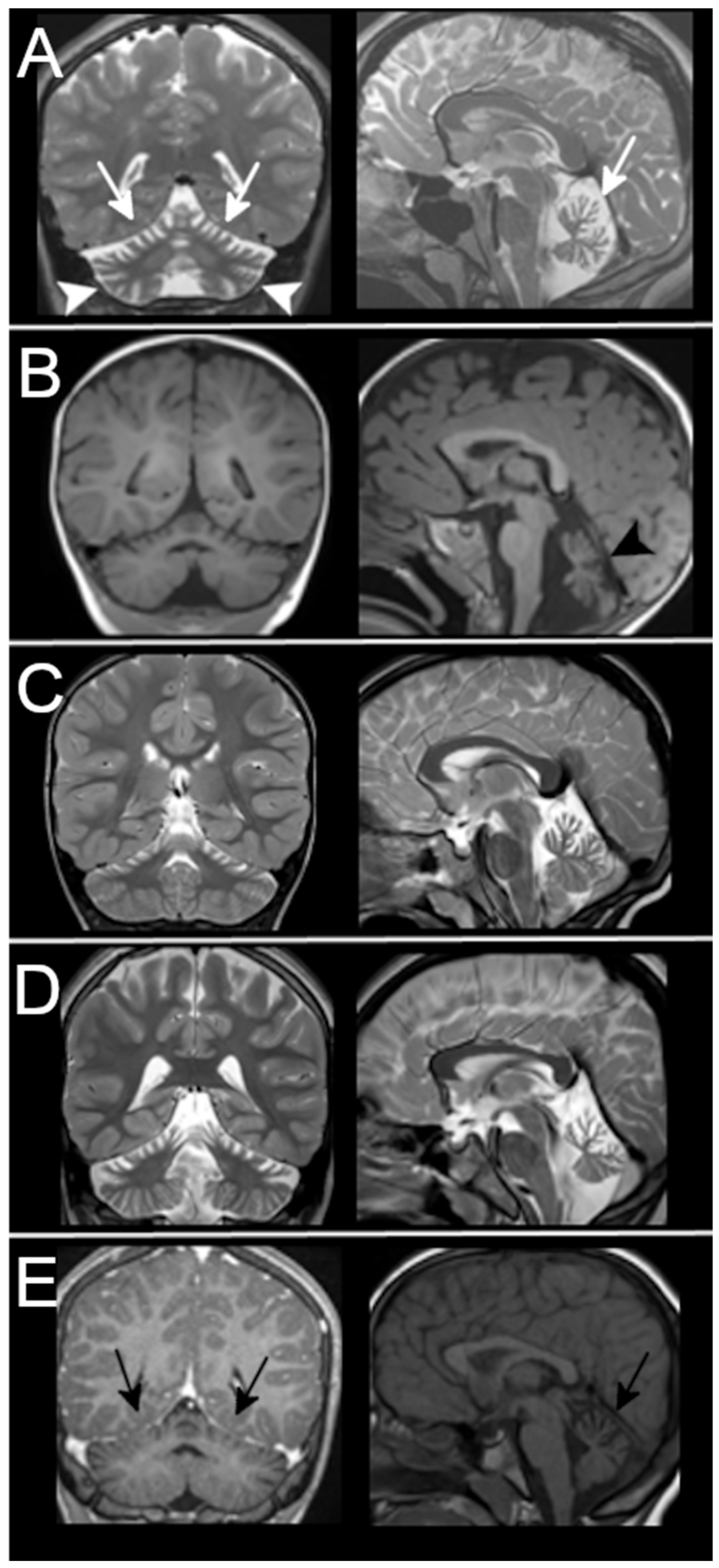
3.3. Neuroradiological Data
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barresi, S.; Niceta, M.; Alfieri, P.; Brankovich, V.; Piccini, G.; Bruselles, A.; Barone, M.; Cusmai, R.; Tartaglia, M.; Bertini, E.; et al. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin. Genet. 2017, 91, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Warman-Chardon, J.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 2012, 7, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, J.; Ali, Z.; Farooq, M.; Khan, K.; Wikström, J.; Iqbal, M.; Zulfiqar, S.; Faryal, S.; Baig, S.M.; Dahl, N. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur. J. Hum. Genet. 2017, 25, 848–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Ohba, C.; Iai, M.; Hirabayashi, S.; Osaka, H.; Hiraide, T.; Saitsu, H.; Matsumoto, N. Sporadic infantile-onset spinocerebellar ataxia caused by missense mutations of the inositol 1,4,5-triphosphate receptor type 1 gene. J. Neurol. 2015, 262, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.J.; Sweeney, M.G.; Li, A.; Treacy, C.; Chandrashekar, H.S.; Giunti, P.; Goold, R.G.; Davis, M.B.; Houlden, H.; Tabrizi, S.J. An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: A genetic, clinical and radiological description. Mov. Disord. 2010, 25, 2176–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambonin, J.L.; Bellomo, A.; Ben-Pazi, H.; Everman, D.B.; Frazer, L.M.; Geraghty, M.T.; Harper, A.D.; Jones, J.R.; Kamien, B.; Kernohan, K.; et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: A case series and review of this emerging congenital ataxia. Orphanet J. Rare Dis. 2017, 12, 121. [Google Scholar] [CrossRef]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Brémond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, T.; Barth, P.; Reneman, L.; Appelhof, B.; Baas, F.; Poll-The, B.T. A de novo missense mutation in the inositol 1,4,5-triphosphate receptor type 1 gene causing severe pontine and cerebellar hypoplasia: Expanding the phenotype of ITPR1-related spinocerebellar ataxia’s. Am. J. Med. Genet. A 2017, 173, 207–212. [Google Scholar] [CrossRef]
- McEntagart, M.; Williamson, K.A.; Rainger, J.K.; Wheeler, A.; Seawright, A.; De Baere, E.; Verdin, H.; Bergendahl, L.T.; Quigley, A.; Rainger, J.; et al. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am. J. Hum. Genet. 2016, 98, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Ohba, C.; Osaka, H.; Iai, M.; Yamashita, S.; Suzuki, Y.; Aida, N.; Shimozawa, N.; Takamura, A.; Doi, H.; Tomita-Katsumoto, A.; et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013, 14, 225–232. [Google Scholar] [CrossRef]
- Dentici, M.L.; Barresi, S.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Bruselles, A.; Pantaleoni, F.; Danieli, A.; Iarossi, G.; Cappa, M.; et al. Identification of novel and hotspot mutations in the channel domain of ITPR1 in two patients with Gillespie syndrome. Gene 2017, 628, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 2008, 71, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Helbig, K.L.; Harmuth, F.; Deconinck, T.; Tanpaiboon, P.; Sun, B.; Guo, W.; Wang, R.; Palmaer, E.; Tang, S.; et al. De novo ITPR1 variants are a recurrent cause of early-onset ataxia, acting via loss of channel function. Eur. J. Hum. Genet. 2018, 26, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadrina, M.I.; Shulskaya, M.V.; Klyushnikov, S.; Nikopensius, T.; Nelis, M.; Kivistik, P.A.; Komar, A.A.; Limborska, S.A.; Illarioshkin, S.N.; A Slominsky, P. ITPR1 gene p.Val1553Met mutation in Russian family with mild Spinocerebellar ataxia. Cerebellum Ataxias 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, E.; Orsi, L.; Godani, M.; Vaula, G.; Jensen, S.; Salmon, E.; Ferrari, G.; Squadrone, S.; Abete, M.C.; Cagnoli, C.; et al. Two Italian Families with ITPR1 Gene Deletion Presenting a Broader Phenotype of SCA15. Cerebellum 2010, 9, 115–123. [Google Scholar] [CrossRef]
- Paganini, L.; Pesenti, C.; Milani, D.; Fontana, L.; Motta, S.; Sirchia, S.M.; Scuvera, G.; Marchisio, P.; Esposito, S.; Cinnante, C.M.; et al. A novel splice site variant in ITPR1 gene underlying recessive Gillespie syndrome. Am. J. Med. Genet. 2018, 176, 1427–1431. [Google Scholar] [CrossRef]
- Wang, L.; Hao, Y.; Yu, P.; Cao, Z.; Zhang, J.; Zhang, X.; Chen, Y.; Zhang, H.; Gu, W. Identification of a Splicing Mutation in ITPR1 via WES in a Chinese Early-Onset Spinocerebellar Ataxia Family. Cerebellum 2018, 17, 294–299. [Google Scholar] [CrossRef] [Green Version]
- Stendel, C.; Wagner, M.; Rudolph, G.; Klopstock, T. Gillespie’s Syndrome with Minor Cerebellar Involvement and No Intellectual Disability Associated with a Novel ITPR1 Mutation: Report of a Case and Literature Review. Neuropediatrics 2019, 50, 382–386. [Google Scholar] [CrossRef]
- Tada, M.; Nishizawa, M.; Onodera, O. Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias. Neurochem. Int. 2016, 94, 1–8. [Google Scholar] [CrossRef]
- Sugawara, T.; Hisatsune, C.; Le, T.D.; Hashikawa, T.; Hirono, M.; Hattori, M.; Nagao, S.; Mikoshiba, K. Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintain-ing the spine morphology of purkinje cells in adult mice. J. Neurosci. 2013, 33, 12186–12196. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, M.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G.; Parys, J.B. Pathophysiological consequences of isoform-specific IP3 receptor mutations. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865 Pt B, 1707–1717. [Google Scholar] [CrossRef]
- Poretti, A.; Wolf, N.; Boltshauser, E. Differential diagnosis of cerebellar atrophy in childhood. Eur. J. Paediatr. Neurol. 2008, 12, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Boltshauser, E.; Poretti, A. Differential Diagnosis of Cerebellar Atrophy in Childhood: An Update. Neuropediatrics 2015, 46, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M.A.; Poskitt, K.J.; Roland, E.H.; Hill, A.; Hendson, G. Cerebellar vermian atrophy after neonatal hypoxic-ischemic en-cephalopathy. AJNR Am. J. Neuroradiol. 2004, 25, 1008–1015. [Google Scholar] [PubMed]
- Brandsma, R.; Verschuuren-Bemelmans, C.; Amrom, D.; Barisic, N.; Baxter, P.; Bertini, E.; Blumkin, L.; Brankovic-Sreckovic, V.; Brouwer, O.; Bürk, K.; et al. A clinical diagnostic algorithm for early onset cerebellar ataxia. Eur. J. Paediatr. Neurol. 2019, 23, 692–706. [Google Scholar] [CrossRef]


{kind=link}
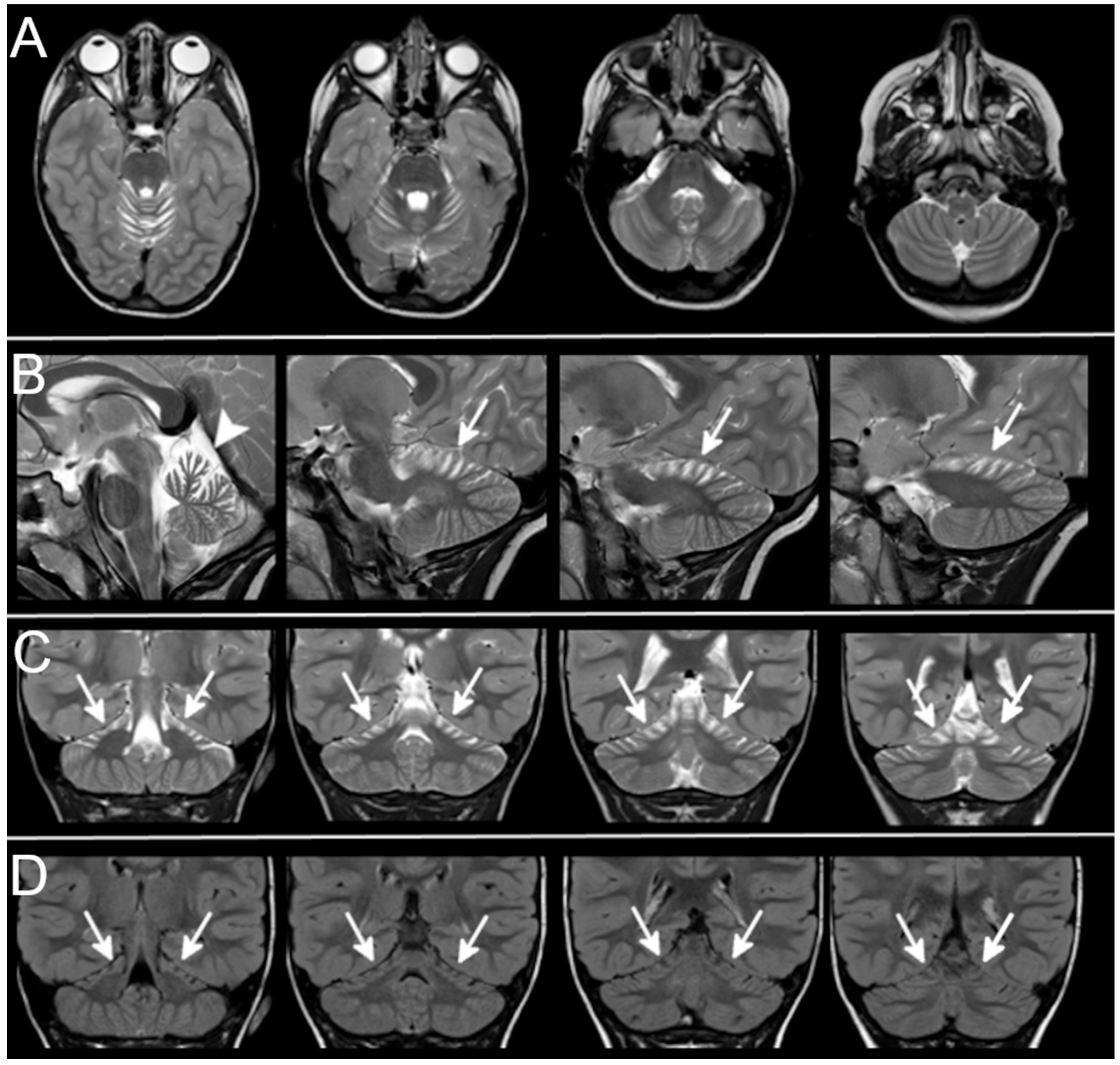{kind=link}
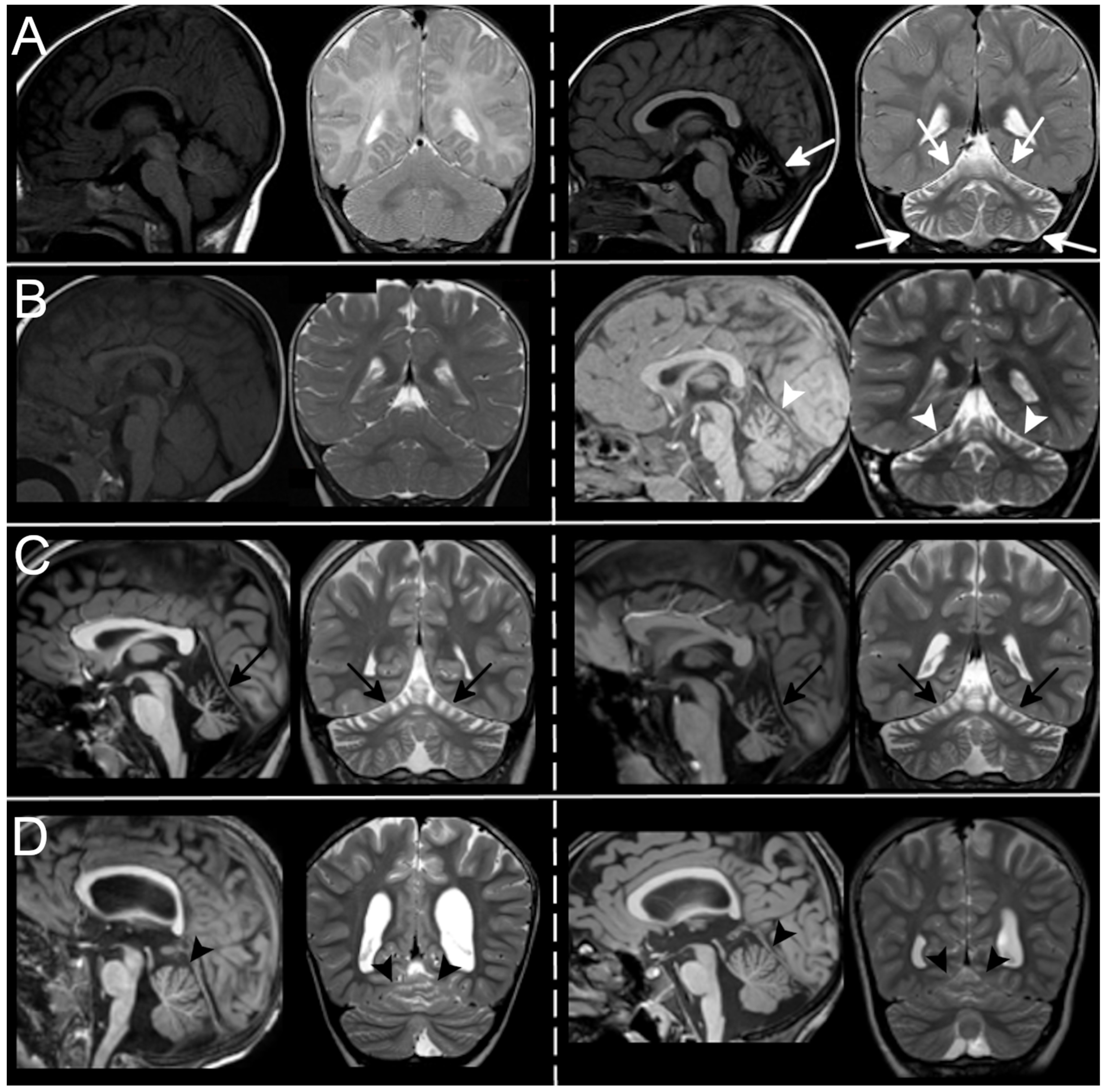{kind=link}
{kind=link}
| Group A | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Family | Patient | Gender; Age at Last Evaluation | Clinical Features Phenotype | Superior Cerebellar Atrophy | Diffuse Atrophy | Progression over Time | Genetics | ACMG Classification | CADD; DANN |
| I | 1. Proband | M; 9 years | DD, ID, hypotonia, ataxia, facial dysmorphisms, cryptorchidism; SCA29 | + | - | No | c2084G > A; p.S695N | Uncertain significance | 20.7; 0.9919 |
| 2. Sister | F; 7 years | DD, ID, hypotonia, ataxia; SCA29 | + | - | NA | ||||
| II | 3. Proband * | M; 28 years | Severe motor delay, normal cognitive level, hypotonia, ataxia, postural tremor, slurred speech, nystagmus, OMA; SCA29 | + | - | NA | c.805C > T; p.R269W | Pathogenic (ClinVar: Pathogenic) [1] | 26.399; 0.9992 |
| 4. Mother * | F; 56 years | Motor delay, normal cognitive level, hypotonia, ataxia, slurred speech, OMA; SCA15 | + | - | NA | ||||
| 5. Brother * | M; 23 years | Severe motor delay, mild ID, hypotonia, ataxia, slurred speech, nystagmus; SCA29 | + | - | NA | ||||
| III | 6. Proband | F; 2 years | Ambulation not achieved, mild ID, hypotonia, ataxia, nystagmus, bilateral iris hypoplasia; Gillespie syndrome | + | - | NA | c.7786-7788delAAG p.K2596del de novo | Pathogenic [9] | // |
| IV | 7. Proband | M; 13 years | DD, mild ID, hypotonia, ataxia, tremor, nystagmus, OMA; SCA29 | - | + | Yes | c.800C > T p.T267M de novo | Pathogenic (ClinVar: Pathogenic) [10] | 26.2; 0.9993 |
| V | 8. Proband * | F; 19 years | Severe motor delay, normal cognitive level, hypotonia, ataxia, postural tremor, slurred speech, nystagmus, OMA; SCA29 | - | + | NA | c.722G > A p.R241K | Pathogenic (ClinVar: Pathogenic) [1] | 28.1; 0.9954 |
| 9. Mother * | F; 42 years | Slurred speech, normal cognitive level; SCA29 | + | - | NA | ||||
| VI | 10. Proband * | F; 29 years | Severe motor delay, moderate ID, hypotonia, ataxia, slurred speech, nystagmus, bilateral iris hypoplasia, ptosis; Gillespie syndrome | - | + | NA | c.7727A > T p.N2576I de novo | Likely pathogenic [11] | 28.5; 0.9913 |
| VII | 11. Proband | M; 12 years | Hypotonia, ataxia, dysarthria, nystagmus, OMA; SCA29 | + | - | NA | c.805C > T; p.R269W de novo | Pathogenic (ClinVar: Pathogenic) [1] | 26.399; 0.9992 |
| VIII | 12. Proband | F; 3 years | DD, hypotonia, ataxia, nystagmus, dysarthria, OMA; SCA29 | + | - | Yes | c.805C > T; p.R269W de novo | Pathogenic (ClinVar: Pathogenic) [1] | 26.399; 0.9992 |
| IX | 13. Proband * | M; 6 years | Moderate motor delay, normal cognitive level, hypotonia, ataxia, postural tremor, slurred speech, nystagmus; SCA29 | + | - | NA | c.839C > A p.A280D de novo | Pathogenic [1] | 28.2; 0.9976 |
| X | 14. Proband * | F; 7 years | Severe motor delay, normal cognitive level, hypotonia, ataxia, postural tremor, slurred speech, nystagmus; SCA29 | + | - | NA | c.1488G > A p.E497K de novo | Likely pathogenic (ClinVar: Likely Pathogenic) [1] | 29.2; 0.9994 |
| Group B | |||||||||
| XI | 15. Proband | F; 6 years | Severe motor delay, ataxia, hypotonia, nystagmus SCA29 | + | - | NA | c.805C > T p.R269W de novo | Pathogenic (ClinVar: Pathogenic) [1] | 26.399; 0.9992 |
| XII | 16. Proband | F; 18 months | Severe motor delay, hypotonia, ataxia, OMA; SCA29 | + | - | NA | c.800C > T p.T267M de novo | Pathogenic (ClinVar: Pathogenic) [10] | 26.2; 0.9993 |
| XIII | 17. Proband | F, 14 years | progressive spastic paraparesis; SCA29 | + | - | NA | negative | // | // |
| XIV | 18. Proband | F, 7 years | Moderate motor delay, moderate ID, hypotonia, ataxia, slurred speech, nystagmus; SCA29 | + | - | NA | c.1489G > A p.E497K de novo | Likely pathogenic (ClinVar: Likely Pathogenic) [1] | 29.2; 0.9994 |
| XV | 19. Proband | M, 12 years | Ambulation not achieved, moderate ID hypotonia, ataxia, slurred speech, nystagmus, OMA; SCA29 | + | - | No | c.800C > T p.T267M de novo | Pathogenic (ClinVar: Pathogenic) [10] | 26.2; 0.9993 |
| Study/Journal | Number of Patients | Age Range | Phenotype | Infratentorial Imaging | Associated Neuroradiological Findings | Progression of Cerebellar Atrophy |
|---|---|---|---|---|---|---|
| Hara et al., 2008 [10] Neurology | 2 families (10 affected members) | NA | SCA15 | Cerebellar atrophy | - | NA |
| Di Gregorio et al., 2010 [11] Cerebellum | 2 families (12 affected members) | 44–81 years | SCA15 (buccolingual dyskinesias, facial myokymias, pyramidal signs) | Cerebellar vermis atrophy with a mild involvement of the hemispheres in some individuals | - | NA |
| Novak et al., 2010 [5] Mov. Disord. | 1 family (3 affected members) | 38–56 years | SCA15 | Moderate cerebellar atrophy, which preferentially involves the superior vermis | Cortical parietal and temporal atrophy | NA |
| Huang et al., 2012 [2] Orphanet J. Rare Dis. | 1 family (3 affected members) | 5–45 years | SCA29 | Mild cerebellar vermis atrophy | - | Yes |
| Sasaki et al., 2015 [4] J. Neurol. | 4 patients | 6–12 years | SCA15, SCA29 | Superior cerebellar hemispheres atrophy, vermian diffuse atrophy | Atrophy of the pontine tegmentum | Yes |
| Gerber et al., 2016 [7] Am. J. Hum. Genet. | 5 patients | 1.5–18 years | Gillespie syndrome | Cerebellar atrophy | Thin CC | Yes |
| Mc Entagart et al., 2016 [9] Am. J. Hum. Genet. | 13 patients | 13–55 years | Gillespie syndrome | Atrophy mainly affecting the superior vermis, involving superior cerebellar hemispheres more than the inferior | Abnormal periventricular increased T2/FLAIR white matter signal adjacent to the frontal horns | Yes |
| Shadrina et al., 2016 [12] Cerebellum Ataxias | 1 family (2 affected members) | 54 years | SCA15 | Mild cerebellar atrophy | - | NA |
| Barresi et al., 2017 [1] Clin. Genet. | 4 families (6 affected members) | 7–28 years | SCA29, SCA15 | Cerebellar and/or vermis atrophy | - | Yes |
| Dentici et al., 2017 [13] Gene | 2 patients | 2–29 years | Gillespie syndrome | Cerebellar atrophy, predominantly in the vermis | - | NA |
| Klar et al., 2017 [3] Eur. J. Hum. Genet. | Family (6 affected members) | 17–45 years | SCA29 | Cerebellar atrophy most pronounced in the vermis | - | NA |
| Van Dijk et al., 2017 [8] Am. J. Med. Genet. | 1 patient | 6 years | SCA29 | The vermis inferior is almost absent and the vermis superior showed hypoplasia with superimposed atrophy. The vermis inferior is almost absent and the vermis superior showed hypoplasia with superimposed atrophy. Almost absent inferior vermis, hypoplasia with atrophy of the superior vermis | Hyperintensities in medulla oblungata | No |
| Zambonin et al., 2017 [6] Orphanet J. Rare Dis. | 21 patients | 28 m–49 years | SCA29 | Cerebellar atrophy, often with superior cerebellar hemispheres and vermis | Pontine atrophy | Yes |
| Paganini et al., 2018 [14] Am. J. Med. Genet. | 1 family (2 affected members) | 6–9 years | Gillespie syndrome | Generalized atrophy, mainly vermis atrophy | - | Yes |
| Synofzik et al., 2018 [15] Eur. J. Hum. Genet. | 5 families (10 affected members) | 33–80 years | SCA15 | Cerebellar atrophy with a major involvement of vermis | - | NA |
| Wang et al., 2018 [16] Cerebellum | 4 patients | 6–51 years | SCA29 | Cerebellar hemisphere atrophy | - | NA |
| Stendel et al., 2019 [17] Neuropediatrics | 1 patient | NA | Gillespie syndrome | Atrophy of the anterior cerebellar vermis | - | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romaniello, R.; Pasca, L.; Panzeri, E.; D’Abrusco, F.; Travaglini, L.; Serpieri, V.; Signorini, S.; Aiello, C.; Bertini, E.; Bassi, M.T.; et al. Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders. Int. J. Mol. Sci. 2022, 23, 6723. https://doi.org/10.3390/ijms23126723
Romaniello R, Pasca L, Panzeri E, D’Abrusco F, Travaglini L, Serpieri V, Signorini S, Aiello C, Bertini E, Bassi MT, et al. Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders. International Journal of Molecular Sciences. 2022; 23(12):6723. https://doi.org/10.3390/ijms23126723
Chicago/Turabian StyleRomaniello, Romina, Ludovica Pasca, Elena Panzeri, Fulvio D’Abrusco, Lorena Travaglini, Valentina Serpieri, Sabrina Signorini, Chiara Aiello, Enrico Bertini, Maria Teresa Bassi, and et al. 2022. "Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders" International Journal of Molecular Sciences 23, no. 12: 6723. https://doi.org/10.3390/ijms23126723
APA StyleRomaniello, R., Pasca, L., Panzeri, E., D’Abrusco, F., Travaglini, L., Serpieri, V., Signorini, S., Aiello, C., Bertini, E., Bassi, M. T., Valente, E. M., Zanni, G., Borgatti, R., & Arrigoni, F. (2022). Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders. International Journal of Molecular Sciences, 23(12), 6723. https://doi.org/10.3390/ijms23126723