
Cross-Talk between the Cytokine IL-37 and Thyroid Hormones in Modulating Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions




Abstract
:1. Introduction—An Association between Chronic Inflammation and Age-Related Metabolic and Vascular Conditions
2. Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions
3. Thyroid Hormone Modes of Action in Aging and Age-Related Diseases
4. The Role of the Cytokine IL-37 in Regulating Acute Inflammation
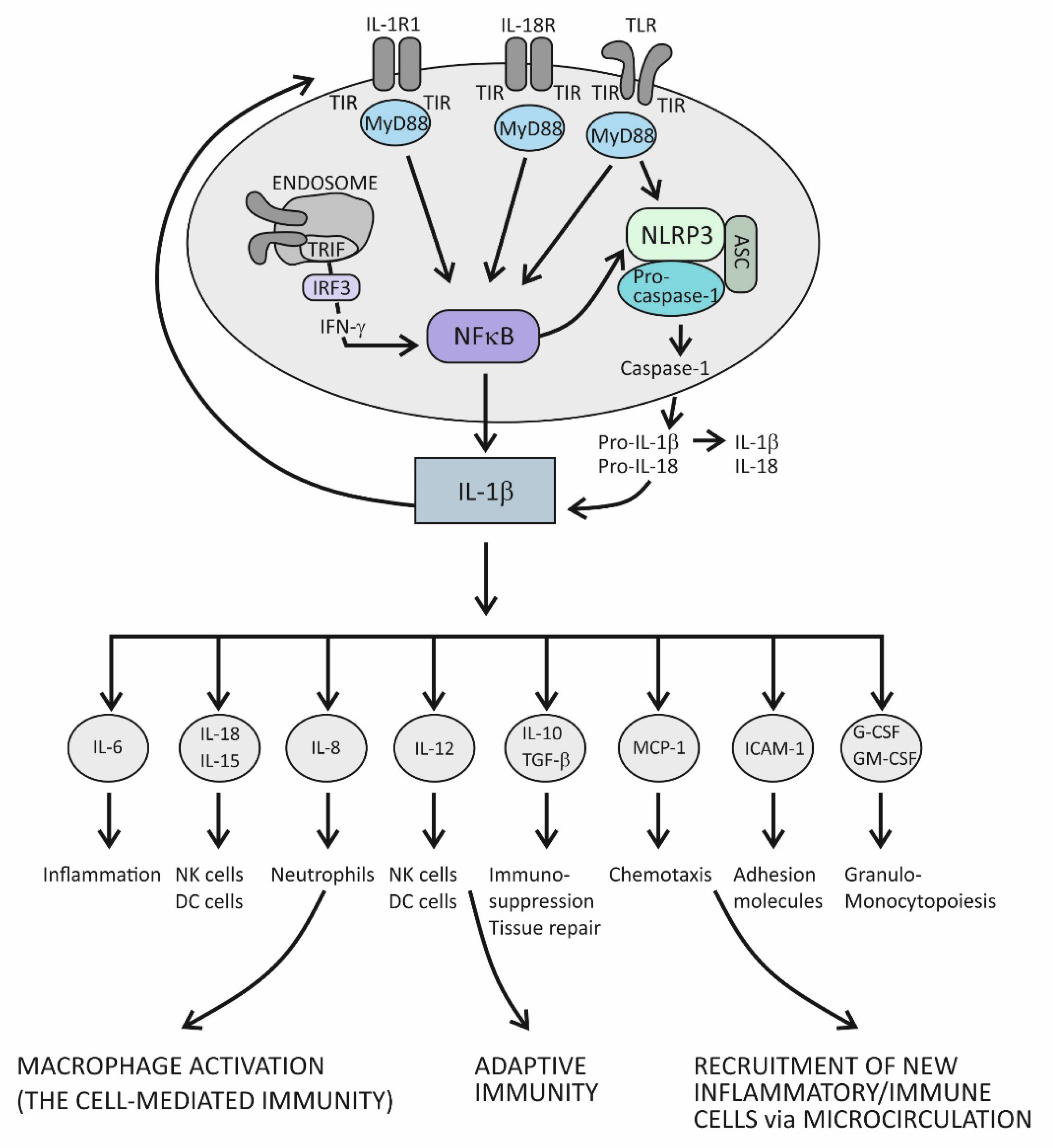
4.1. Innate Immunity and Acute Inflammation
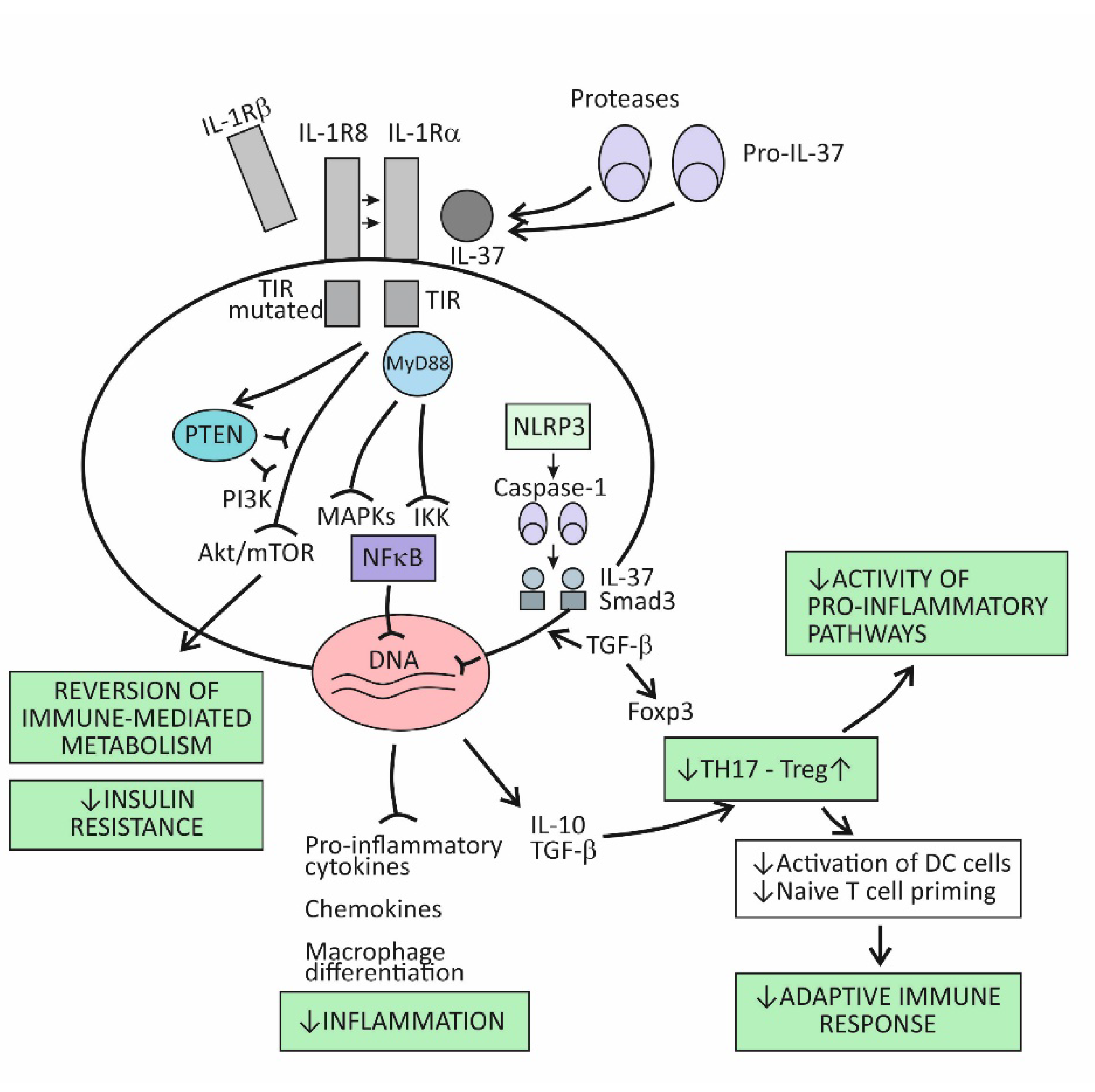
4.2. Suppression of Acute Inflammation by the Cytokine IL-37
5. The Cytokine IL-37 as the Key Regulator of Chronic Inflammation Associated with Organ Damage in Age-Related Metabolic and Vascular Conditions
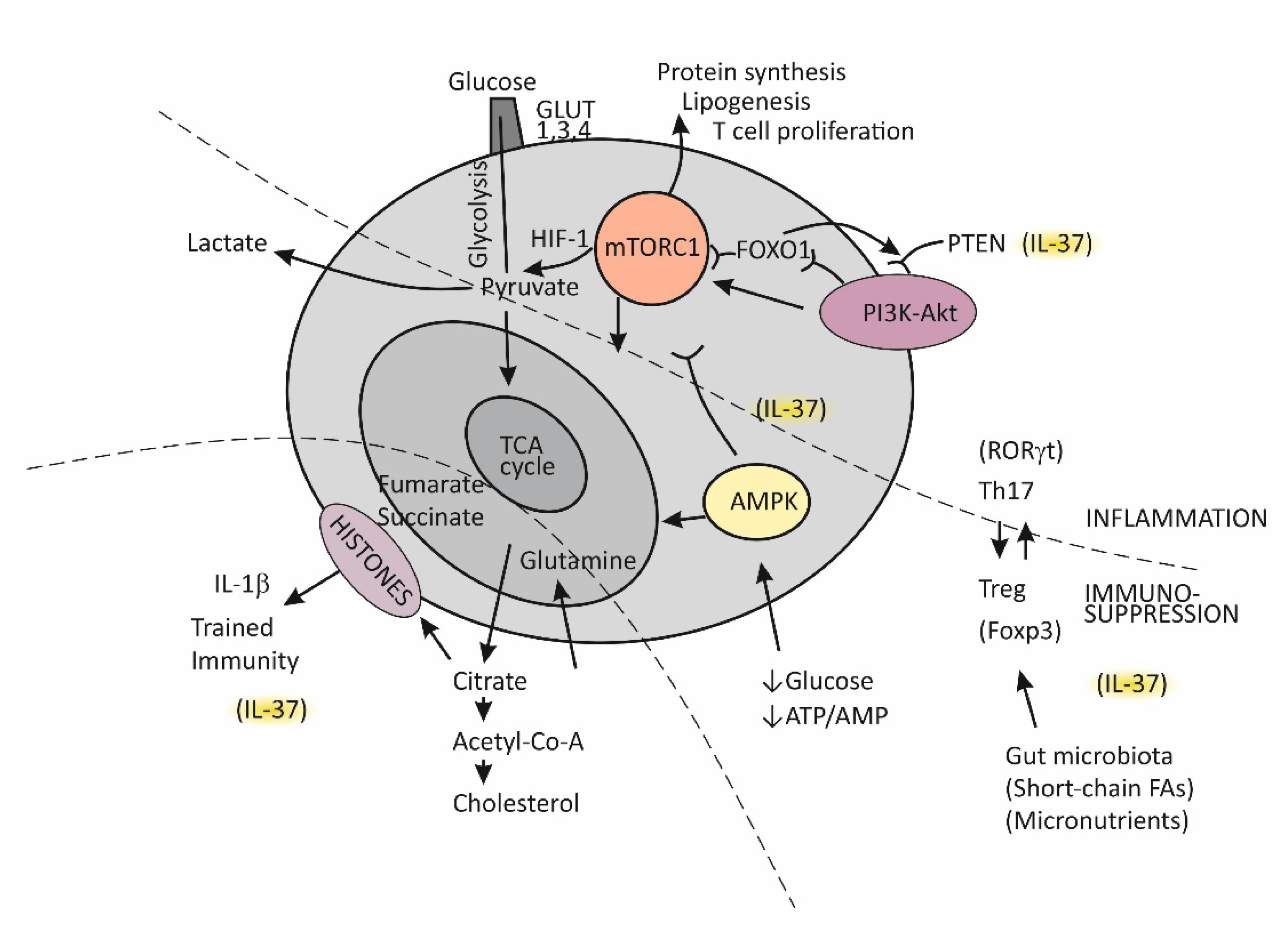
5.1. Immune-Metabolic Disturbances in Age-Related Metabolic and Vascular Conditions
5.2. Trained Immunity
5.3. The Complex Molecule mTOR-Mediated Regulation of the Effector T-Cell Commitment
5.4. Insulin Resistance
5.5. The Role of the Cytokine IL-37 in Reversing Immune-Metabolic Disturbances Associated with Chronic Inflammation
6. Cross-Talk between the Cytokine IL-37 and Thyroid Hormones—A Relationship between Tissue-Related and Whole-Body Responses to Stress
7. Markers of Target Organ Damage can Contribute to Risk Stratification of Older Individuals with Metabolic and Vascular Comorbidities
8. The Model of Target Organ Damage in Older Patients with T2D Based on Patterns of Serum TSH and IL-37
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medzhitov, R. The spectrum of inflammatory responses. Science 2021, 374, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute Inflammation and Metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Uciechowski, P.; Dempke, W. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef]
- Venet, F.; Monneret, G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018, 14, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Zefferino, R.; Di Gioia, S.; Conese, M. Molecular links between endocrine, nervous and immune system during chronic stress. Brain Behav. 2021, 11, e01960. [Google Scholar] [CrossRef]
- Clerencia-Sierra, M.; Ioakeim-Skoufa, I.; Poblador-Plou, B.; González-Rubio, F.; Aza-Pascual-Salcedo, M.; Gimeno-Miguel, M.; Prados-Torres, A. Do Centenarians Die Healthier than Younger Elders? A Comparative Epidemiological Study in Spain. J. Clin. Med. 2020, 9, 1563. [Google Scholar] [CrossRef]
- Prajapati, B.; Jena, P.K.; Rajput, P.; Purandhar, K.; Seshadri, S. Understanding and modulating the Toll like Receptors (TLRs) and NOD like Receptors (NLRs) cross talk in type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Antony, V.; Wang, Y.; Wu, G.; Liang, G. Pattern recognition receptor-mediated inflammation in diabetic vascular complications. Med. Res. Rev. 2020, 40, 2466–2484. [Google Scholar] [CrossRef] [PubMed]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Picu, A.; Petcu, L.; Cucu, N.; Chifiriuc, M.C. Gut Microbiota, Host Organism, and Diet Trialogue in Diabetes and Obesity. Front. Nutr. 2019, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepehri, Z.; Kiani, Z.; Afshari, M.; Kohan, F.; Dalvand, A.; Ghavami, S. Inflammasomes and type 2 diabetes: An updated systematic review. Immunol. Lett. 2017, 192, 97–103. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Chu, C.; Artis, D.; Chiu, I.M. Neuro-immune Interactions in the Tissues. Immunity 2020, 52, 464–474. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Trtica Majnarić, L.; Bosnić, Z.; Kurevija, T.; Wittlinger, T. Cardiovascular risk and aging: The need for a more comprehensive understanding. J. Geriatr. Cardiol. JGC 2021, 18, 462–478. [Google Scholar]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Legiawati, L. The Role of Oxidative Stress, Inflammation, and Advanced Glycation End Product in Skin Manifestations of Diabetes Mellitus. Curr. Diabetes Rev. 2022, 18, 87–92. [Google Scholar] [CrossRef]
- Litak, J.; Mazurek, M.; Kulesza, B.; Szmygin, P.; Litak, J.; Kamieniak, P.; Grochowski, C. Cerebral Small Vessel Disease. Int. J. Mol. Sci. 2020, 21, 9729. [Google Scholar] [CrossRef] [PubMed]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef] [PubMed]
- Climie, R.E.; van Sloten, T.T.; Bruno, R.M.; Taddei, S.; Empana, J.P.; Stehouwer, C.; Sharman, J.E.; Boutouyrie, P.; Laurent, S. Macrovasculature and Microvasculature at the Crossroads Between Type 2 Diabetes Mellitus and Hypertension. Hypertension 2019, 73, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Sica, V.; Kroemer, G. IMMP2L: A mitochondrial protease suppressing cellular senescence. Cell Res. 2018, 28, 607–608. [Google Scholar] [CrossRef]
- Strain, W.D.; Paldánius, P.M. Correction to: Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2021, 20, 120. [Google Scholar] [CrossRef]
- Tomiyama, H.; Shiina, K.; Matsumoto-Nakano, C.; Ninomiya, T.; Komatsu, S.; Kimura, K.; Chikamori, T.; Yamashina, A. The Contribution of Inflammation to the Development of Hypertension Mediated by Increased Arterial Stiffness. J. Am. Heart Assoc. 2017, 6, e005729. [Google Scholar] [CrossRef]
- Trtica Majnarić, L.; Guljaš, S.; Bosnić, Z.; Šerić, V.; Wittlinger, T. Neutrophil-to-Lymphocyte Ratio as a Cardiovascular Risk Marker May Be Less Efficient in Women Than in Men. Biomolecules 2021, 11, 528. [Google Scholar] [CrossRef]
- Chang, Y.C.; Hee, S.W.; Chuang, L.M. T helper 17 cells: A new actor on the stage of type 2 diabetes and aging? J. Diabetes Investig. 2021, 12, 909–913. [Google Scholar] [CrossRef]
- Zhang, S.; Gang, X.; Yang, S.; Cui, M.; Sun, L.; Li, Z.; Wang, G. The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Front. Immunol. 2021, 12, 678355. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Araos, P.; Figueroa, S.; Amador, C.A. The Role of Neutrophils in Hypertension. Int. J. Mol. Sci. 2020, 21, 8536. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef]
- Brown, C.Y.; Sadlon, T.; Hope, C.M.; Wong, Y.Y.; Wong, S.; Liu, N.; Withers, H.; Brown, K.; Bandara, V.; Gundsambuu, B.; et al. Molecular Insights into Regulatory T-Cell Adaptation to Self, Environment, and Host Tissues: Plasticity or Loss of Function in Autoimmune Disease. Front. Immunol. 2020, 11, 1269. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Ip, B.; Cilfone, N.A.; Belkina, A.C.; DeFuria, J.; Jagannathan-Bogdan, M.; Zhu, M.; Kuchibhatla, R.; McDonnell, M.E.; Xiao, Q.; Kepler, T.B.; et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity 2016, 24, 102–112. [Google Scholar] [CrossRef]
- Bianco, A.C.; Dumitrescu, A.; Gereben, B.; Ribeiro, M.O.; Fonseca, T.L.; Fernandes, G.W.; Bocco, B. Paradigms of Dynamic Control of Thyroid Hormone Signaling. Endocr. Rev. 2019, 40, 1000–1047. [Google Scholar] [CrossRef]
- Cicatiello, A.G.; Di Girolamo, D.; Dentice, M. Metabolic Effects of the Intracellular Regulation of Thyroid Hormone: Old Players, New Concepts. Front. Endocrinol. 2018, 9, 474. [Google Scholar] [CrossRef]
- Lourbopoulos, A.I.; Mourouzis, I.S.; Trikas, A.G.; Tseti, I.K.; Pantos, C.I. Effects of Thyroid Hormone on Tissue Hypoxia: Relevance to Sepsis Therapy. J. Clin. Med. 2021, 10, 5855. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, bnz008. [Google Scholar] [CrossRef] [PubMed]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. International journal of molecular sciences. Int. J. Mol. Sci. 2020, 21, 4140. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, K.; Billon, C.; Bissler, M.; Beylot, M.; Lobaccaro, J.M.; Vanacker, J.M.; Samarut, J. Thyroid hormone receptor beta (TRbeta) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 2010, 285, 28156–28163. [Google Scholar] [CrossRef] [Green Version]
- Bomer, N.; Pavez-Giani, M.G.; Deiman, F.E.; Linders, A.N.; Hoes, M.F.; Baierl, C.; Oberdorf-Maass, S.U.; de Boer, R.A.; Silljé, H.; Berezikov, E.; et al. Selenoprotein DIO2 Is a Regulator of Mitochondrial Function, Morphology and UPRmt in Human Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 11906. [Google Scholar] [CrossRef]
- Mendez, D.A.; Ortiz, R.M. Thyroid hormones and the potential for regulating glucose metabolism in cardiomyocytes during insulin resistance and T2DM. Physiol. Rep. 2021, 9, e14858. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int. J. Mol. Sci. 2017, 18, 744. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, A.; García-Carpizo, V.; Gallardo, M.E.; Villamuera, R.; Gómez-Ferrería, M.A.; Pascual, A.; Buisine, N.; Sachs, L.M.; Garesse, R.; Aranda, A. The thyroid hormone receptor β induces DNA damage and premature senescence. J. Cell Biol. 2014, 204, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Coulis, G.; Londhe, A.D.; Sagabala, R.S.; Shi, Y.; Labbé, D.P.; Bergeron, A.; Sahadevan, P.; Nawaito, S.A.; Sahmi, F.; Josse, M.; et al. Protein tyrosine phosphatase 1B regulates miR-208b-argonaute 2 association and thyroid hormone responsiveness in cardiac hypertrophy. Sci. Signal. 2022, 15, eabn6875. [Google Scholar] [CrossRef]
- Femia, M.R.; Evans, R.M.; Zhang, J.; Sun, X.; Lebegue, C.J.; Roggero, V.R.; Allison, L.A. Mediator subunit MED1 modulates intranuclear dynamics of the thyroid hormone receptor. J. Cell. Biochem. 2020, 121, 2909–2926. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.C.; Kuo, H.Y.; Hong, H.K.; Cedernaes, J.; Hepler, C.; Wright, A.G.; Sommars, M.A.; Kobayashi, Y.; Marcheva, B.; Gao, P.; et al. NADH inhibition of SIRT1 links energy state to transcription during time-restricted feeding. Nat. Metab. 2021, 3, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Kinney, C.J.; Bloch, R.J. µ-Crystallin: A thyroid hormone binding protein. Endocr. Regul. 2021, 55, 89–102. [Google Scholar] [CrossRef]
- Davis, P.J.; Mousa, S.A.; Lin, H.Y. Nongenomic Actions of Thyroid Hormone: The Integrin Component. Physiol. Rev. 2021, 101, 319–352. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Yen, P.M. Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone. Int. J. Mol. Sci. 2018, 19, 3284. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Teixeira, P.; Dos Santos, P.B.; Pazos-Moura, C.C. The role of thyroid hormone in metabolism and metabolic syndrome. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820917869. [Google Scholar] [CrossRef]
- Bano, A.; Chaker, L.; Muka, T.; Mattace-Raso, F.; Bally, L.; Franco, O.H.; Peeters, R.P.; Razvi, S. Thyroid Function and the Risk of Fibrosis of the Liver, Heart, and Lung in Humans: A Systematic Review and Meta-Analysis. Thyroid. Off. J. Am. Thyroid. Assoc. 2020, 30, 806–820. [Google Scholar] [CrossRef]
- De Luca, R.; Davis, P.J.; Lin, H.Y.; Gionfra, F.; Percario, Z.A.; Affabris, E.; Pedersen, J.Z.; Marchese, C.; Trivedi, P.; Anastasiadou, E.; et al. Thyroid Hormones Interaction with Immune Response, Inflammation and Non-Thyroidal Illness Syndrome. Front. Cell Dev. Biol. 2021, 8, 614030. [Google Scholar] [CrossRef]
- Fülöp, T.; Larbi, A.; Witkowski, J.M. Human Inflammaging. Gerontology 2019, 65, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bekassy, Z.; Lopatko Fagerström, I.; Bader, M.; Karpman, D. Crosstalk between the renin-angiotensin, complement and kallikrein-kinin systems in inflammation. Nat. Rev. Immunol. 2021, 1–18. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Ogawa-Wong, A.; Carmody, C.; Ambrosio, R.; Cicatiello, A.G.; Luongo, C.; Salvatore, D.; Handy, D.E.; Larsen, P.R.; Wajner, S.M.; et al. A Type 2 Deiodinase-Dependent Increase in Vegfa Mediates Myoblast-Endothelial Cell Crosstalk During Skeletal Muscle Regeneration. Thyroid. Off. J. Am. Thyroid. Assoc. 2021, 31, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Khor, S. “Hypothalamic Microinflammation” Paradigm in Aging and Metabolic Diseases. Cell Metab. 2019, 30, 19–35. [Google Scholar] [CrossRef]
- Bolborea, M.; Langlet, F. What is the physiological role of hypothalamic tanycytes in metabolism? Am. J. Physiology. Regul. Integr. Comp. Physiol. 2021, 320, R994–R1003. [Google Scholar] [CrossRef]
- Mu, W.; Li, S.; Xu, J.; Guo, X.; Wu, H.; Chen, Z.; Qiao, L.; Helfer, G.; Lu, F.; Liu, C.; et al. Hypothalamic Rax+ tanycytes contribute to tissue repair and tumorigenesis upon oncogene activation in mice. Nat. Commun. 2021, 12, 2288. [Google Scholar] [CrossRef]
- Dorrier, C.E.; Jones, H.E.; Pintarić, L.; Siegenthaler, J.A.; Daneman, R. Emerging roles for CNS fibroblasts in health, injury and disease. Nat. Rev. Neurosci. 2022, 23, 23–34. [Google Scholar] [CrossRef]
- Frenzel, A.; Binder, H.; Walter, N.; Wirkner, K.; Loeffler, M.; Loeffler-Wirth, H. The aging human body shape. NPJ Aging Mech. Dis. 2020, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Wilmanski, T.; Diener, C.; Rappaport, N.; Patwardhan, S.; Wiedrick, J.; Lapidus, J.; Earls, J.C.; Zimmer, A.; Glusman, G.; Robinson, M.; et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 2021, 3, 274–286. [Google Scholar] [CrossRef]
- Walczak, K.; Sieminska, L. Obesity and Thyroid Axis. Int. J. Environ. Res. Public Health 2021, 18, 9434. [Google Scholar] [CrossRef]
- Papadopoulou, A.M.; Bakogiannis, N.; Skrapari, I.; Moris, D.; Bakoyiannis, C. Thyroid Dysfunction and Atherosclerosis: A Systematic Review. In Vivo 2020, 34, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Barajon, I.; Mantovani, A.; Garlanda, C. Regulatory Role of IL-1R8 in Immunity and Disease. Front. Immunol. 2016, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Introduction to the interleukin-1 family of cytokines and receptors: Drivers of innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Orning, P.; Lien, E.; Fitzgerald, K.A. Gasdermins and their role in immunity and inflammation. J. Exp. Med. 2019, 216, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Rex, D.; Agarwal, N.; Prasad, T.; Kandasamy, R.K.; Subbannayya, Y.; Pinto, S.M. A comprehensive pathway map of IL-18-mediated signalling. J. Cell Commun. Signal. 2020, 14, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Uzhachenko, R.V.; Shanker, A. CD8+ T Lymphocyte and NK Cell Network: Circuitry in the Cytotoxic Domain of Immunity. Front. Immunol. 2019, 10, 1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Haan, J.M.; Arens, R.; van Zelm, M.C. The activation of the adaptive immune system: Cross-talk between antigen-presenting cells, T cells and B cells. Immunol. Lett. 2014, 162, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Quan, Y.; Yue, Y.; Heng, X.; Che, F. Interleukin-37: A crucial cytokine with multiple roles in disease and potentially clinical therapy. Oncol. Lett. 2018, 15, 4711–4719. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Xie, B.; Wu, G.; Hu, J.; Wang, D.; Cai, X.; Li, J. Interleukin-37: The Effect of Anti-Inflammatory Response in Human Coronary Artery Endothelial Cells. Mediat. Inflamm. 2019, 2019, 2650590. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef] [Green Version]
- Walls, J.; Sinclair, L.; Finlay, D. Nutrient sensing, signal transduction and immune responses. Semin. Immunol. 2016, 28, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.; Joosten, L.A.; Netea, M.G. Immunometabolic circuits in trained immunity. Semin. Immunol. 2016, 28, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Luo, W.; Han, J.; Khan, Z.A.; Fang, Q.; Jin, Y.; Chen, X.; Zhang, Y.; Wang, M.; Qian, J.; et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat. Commun. 2020, 11, 2148. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.; Glastras, S.J.; Wong, M.Y.W.; Pollock, C.A.; Saad, S. The Role of the Gut Microbiome in Diabetes and Obesity-Related Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9641. [Google Scholar] [CrossRef] [PubMed]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Bekkering, S.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Trained innate immunity and atherosclerosis. Curr. Opin. Lipidol. 2013, 24, 487–492. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [Green Version]
- Salmond, R.J. mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Barbi, J.; Pardoll, D.; Pan, F. Metabolic control of the Treg/Th17 axis. Immunol. Rev. 2013, 252, 52–77. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couple immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, K.; Akash, M.S. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatric Diabetes 2019, 20, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Chang, Y.W.; Hung, L.C.; Chen, Y.C.; Wang, W.H.; Lin, C.Y.; Tzeng, H.H.; Suen, J.L.; Chen, Y.H. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 2021, 11, 587229. [Google Scholar] [CrossRef]
- Sebastian-Valverde, M.; Pasinetti, G.M. The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells 2020, 9, 1552. [Google Scholar] [CrossRef]
- Cavalli, G.; Tengesdal, I.W.; Gresnigt, M.; Nemkov, T.; Arts, R.; Domínguez-Andrés, J.; Molteni, R.; Stefanoni, D.; Cantoni, E.; Cassina, L.; et al. The anti-inflammatory cytokine interleukin-37 is an inhibitor of trained immunity. Cell Rep. 2021, 35, 108955. [Google Scholar] [CrossRef]
- Hamilton, J.; Lee, M.Y.; Hunter, R.; Ank, R.S.; Story, J.Y.; Talekar, G.; Sisroe, T.; Ballak, D.B.; Fedanov, A.; Porter, C.C.; et al. Interleukin-37 improves T-cell-mediated immunity and chimeric antigen receptor T-cell therapy in aged backgrounds. Aging Cell 2021, 20, e13309. [Google Scholar] [CrossRef]
- Cavalli, G.; Justice, J.N.; Boyle, K.E.; D’Alessandro, A.; Eisenmesser, E.Z.; Herrera, J.J.; Hansen, K.C.; Nemkov, T.; Stienstra, R.; Garlanda, C.; et al. Interleukin 37 reverses the metabolic cost of inflammation, increases oxidative respiration, and improves exercise tolerance. Proc. Natl. Acad. Sci. USA 2017, 114, 2313–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, H.; Li, W.; Chen, S.; Feng, T.; Jiao, W.; Wu, C.; Dong, J.; Li, Y.; Li, S.; et al. Interleukin-37 sensitize the elderly type 2 diabetic patients to insulin therapy through suppressing the gut microbiota dysbiosis. Mol. Immunol. 2019, 112, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Meng, S.; Song, R.H.; Qin, Q.; Wang, X.; Yao, Q.; Jiang, Y.; Jiang, W.; Shi, L.; Xu, J.; et al. Polymorphism of IL37 gene as a protective factor for autoimmune thyroid disease. J. Mol. Endocrinol. 2015, 55, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Baek, A.R.; Lee, J.H.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. IL-37 Attenuates Lung Fibrosis by Inducing Autophagy and Regulating TGF-β1 Production in Mice. J. Immunol. 2019, 203, 2265–2275. [Google Scholar] [CrossRef]
- Allaire, J.M.; Poon, A.; Crowley, S.M.; Han, X.; Sharafian, Z.; Moore, N.; Stahl, M.; Bressler, B.; Lavoie, P.M.; Jacobson, K.; et al. Interleukin-37 regulates innate immune signaling in human and mouse colonic organoids. Sci. Rep. 2021, 11, 8206. [Google Scholar] [CrossRef]
- Rakov, H.; De Angelis, M.; Renko, K.; Hönes, G.S.; Zwanziger, D.; Moeller, L.C.; Schramm, K.W.; Führer, D. Aging Is Associated with Low Thyroid State and Organ-Specific Sensitivity to Thyroxine. Thyroid 2019, 29, 1723–1733. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Niccolai, F.; Pasqualetti, G.; Tognini, S.; Magno, S.; Riccioni, T.; Bottari, M.; Caraccio, N.; Monzani, F. Hypothyroidism in the Elderly: Who Should Be Treated and How? J. Endocr. Soc. 2019, 3, 146–158. [Google Scholar] [CrossRef]
- Leng, O.; Razvi, S. Hypothyroidism in the older population. Thyroid. Res. 2019, 12, 2. [Google Scholar] [CrossRef]
- Calsolaro, V.; Niccolai, F.; Pasqualetti, G.; Calabrese, A.M.; Polini, A.; Okoye, C.; Magno, S.; Caraccio, N.; Monzani, F. Overt and Subclinical Hypothyroidism in the Elderly: When to Treat? Front. Endocrinol 2019, 10, 177. [Google Scholar] [CrossRef]
- Lupoli, R.; Di Minno, A.; Tortora, A.; Ambrosino, P.; Lupoli, G.A.; Di Minno, M.N.D. Effects of Treatment with Metformin on TSH Levels: A Meta-analysis of Literature Studies. J. Clin. Endocrinol. Metab. 2014, 99, E143–E148. [Google Scholar] [CrossRef] [Green Version]
- Mourits, V.P.; Helder, L.S.; Matzaraki, V.; Koeken, V.A.C.M.; Groh, L.; de Bree, L.C.J.; Moorlag, S.J.C.F.M.; van der Heijden, C.D.C.C.; Keating, S.T.; van Puffelen, J.H.; et al. The role of sirtuin 1 on the induction of trained immunity. Cell Immunol 2021, 366, 104393. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Mims, B.M.; Castro-Piedras, I.; Barnes, B.J.; Grisham, M.B.; Rahman, R.L.; Pruitt, K. The Versatility of Sirtuin-1 in Endocrinology and Immunology. Front. Cell Dev. Biol. 2020, 8, 589016. [Google Scholar] [CrossRef]
- Galow, A.M.; Peleg, S. How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span. Cells 2022, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.W.; Roelfsema, F.; van der Spoel, E.; Akintola, A.A.; Postmus, I.; Ballieux, B.E.; Slagboom, P.E.; Cobbaert, C.M.; van der Grond, J.; Westendorp, R.G.; et al. Familial Longevity Is Associated with Higher TSH Secretion and Strong TSH-fT3 Relationship. J. Clin. Endocrinol. Metab. 2015, 100, 3806–3813. [Google Scholar] [CrossRef] [Green Version]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Ren. Physiol. 2016, 311, 1087–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, S.; Van der Graaf, Y.; Cramer, M.J.; Kapelle, L.J.; de Borst, G.J.; Visseren, F.; Westerink, J.; SMART study group. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc. Diabetol. 2021, 20, 220. [Google Scholar] [CrossRef] [PubMed]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr. Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Bellary, S.; Kyrou, I.; Brown, J.E.; Bailey, C.J. Type 2 diabetes mellitus in older adults: Clinical considerations and management. Nat. Rev. Endocrinol. 2021, 17, 534–548. [Google Scholar] [CrossRef]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Lee, M.Y.Y.; Bahl, V.; Traum, D.; Schug, J.; Kusmartseva, I.; Atkinson, M.A.; Fan, G.; HPAP Consortium; Kaestner, K.H. Single-cell analysis of the human pancreas in type 2 diabetes using multi-spectral imaging mass cytometry. Cell Rep. 2021, 37, 109919. [Google Scholar] [CrossRef]
- Arababadi, M.K.; Nosratabadi, R.; Hassanshahi, G.; Yaghini, N.; Pooladvand, V.; Shamsizadeh, A.; Hakimi, H.; Derakhshan, R. Nephropathic complication of type-2 diabetes is following pattern of autoimmune diseases? Diabetes Res. Clin. Pract. 2010, 87, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Ramírez, B.; Rodríguez, A.; Becerril, S.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G.; et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell. Mol. Immunol. 2021, 18, 1045–1057. [Google Scholar] [CrossRef]
- Qiu, A.W.; Cao, X.; Zhang, W.W.; Liu, Q.H. IL-17A is involved in diabetic inflammatory pathogenesis by its receptor IL-17RA. Exp. Biol. Med. 2021, 246, 57–65. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Zhou, Y.; Fei, B. Interleukin 37 (IL-37) Reduces High Glucose-Induced Inflammation, Oxidative Stress, and Apoptosis of Podocytes by Inhibiting the STAT3-Cyclophilin A (CypA) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e922979. [Google Scholar] [CrossRef]
- Zoungas, S.; Woodward, M.; Li, Q.; Cooper, M.E.; Hamet, P.; Harrap, S.; Heller, S.; Marre, M.; Patel, A.; Poulter, N.; et al. ADVANCE Collaborative group. Impact of age, age at diagnosis and duration of diabetes on the risk of macrovascular and microvascular complications and death in type 2 diabetes. Diabetologia 2014, 57, 2465–2474. [Google Scholar] [CrossRef]
- Trtica Majnarić, L.; Martinović, I.; Šabanović, Š.; Rudan, S.; Babič, F.; Wittlinger, T. The Effect of Hypertension Duration and the Age of Onset on CV Risk Factors Expression in Perimenopausal Women. Int. J. Hypertens. 2019, 2019, 9848125. [Google Scholar] [CrossRef]
- Nowakowska, M.; Zghebi, S.S.; Ashcroft, D.M.; Buchan, I.; Chew-Graham, C.; Holt, T.; Mallen, C.; van Marwijk, H.; Peek, N.; Perera-Salazar, R.; et al. The comorbidity burden of type 2 diabetes mellitus: Patterns, clusters and predictions from a large English primary care cohort. BMC Med. 2019, 17, 145. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Brorsson, C.A.; Bizzotto, R.; Mari, A.; Tura, A.; Koivula, R.; Mahajan, A.; Vinuela, A.; Tajes, J.F.; Sharma, S.; et al. Four groups of type 2 diabetes contribute to the etiological and clinical heterogeneity in newly diagnosed individuals: An IMI DIRECT study. Cell Rep. Med. 2022, 3, 100477. [Google Scholar] [CrossRef]
- Majnarić, L.T.; Bekić, S.; Babič, F.; Pusztová, Ľ.; Paralič, J. Cluster Analysis of the Associations among Physical Frailty, Cognitive Impairment and Mental Disorders. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e924281. [Google Scholar] [CrossRef]
- Bekić, S.; Babič, F.; Pavlišková, V.; Paralič, J.; Wittlinger, T.; Majnarić, L.T. Clusters of Physical Frailty and Cognitive Impairment and Their Associated Comorbidities in Older Primary Care Patients. Healthcare 2021, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Bosnic, Z.; Yildirim, P.; Babič, F.; Šahinović, I.; Wittlinger, T.; Martinović, I.; Majnaric, L.T. Clustering Inflammatory Markers with Sociodemographic and Clinical Characteristics of Patients with Diabetes Type 2 Can Support Family Physicians’ Clinical Reasoning by Reducing Patients’ Complexity. Healthcare 2021, 9, 1687. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Phenotype | Inflammation- and Metabolic-Related Changes | Patterns of Serum TSH (Min–Max 0.15–9.45) (Mean 2.91) (mU/L) and IL-37 (Min–Max 0.14–258.80) (one extreme value excluded), IQR (21.2–48.0) (Median 13.4) |
|---|---|---|
| Slow-rate progression of organ damage Low-level system Inflammation—low-level insulin resistance | Low-rate migration of inflammatory/immune cells into tissues Low stimulus for IL-37 induction Low-level insulin resistance | Low IL-37 (0.24) TSH normal (2.1) |
| Rapid-rate progression of organ damage but low-level organ damage Low-level system inflammation—low-level insulin resistance | Rapid migration of inflammatory/ immune cells into tissue Moderate tissue inflammation overcomes tissue damage Inflammation/metabolic changes maintained at the tissue level | Higher IL-37 (10.2)—can compensate for metabolic cost of tissue inflammation TSH normal (2.2) |
| Slow-rate progression of organ damage Medium-level organ damage Higher-level system inflammation—higher-level insulin resistance | Low rate of inflammatory/immune cell migration into tissues Tissue repair processes succeed to reverse tissue homeostasis Low stimulus for IL-37 induction The need for TSH to overcome metabolic costs of increased system inflammation | Low IL-37 (0.22) TSH higher (2.39) |
| High-level organ damage High-level system inflammation—high-level insulin resistance | Low-rate inflammatory/immune cell migration into tissues—low tissue receptivity for new cells High-level tissue inflammation— high-level organ damage (fibrosis)— repair and remodeling processes Balanced with inflammatory processes | High IL-37 (16.4) (the need to ameliorate tissue fibrotic processes and to compensate for metabolic costs of inflammation) (1) TSH high (3.3) (the need to compensate for high-level insulin resistance) (2) TSH normal (hypothetically) (tissue resistance to thyroid hormones—the HPT axis is broken down) |
| Moderate-level organ damage Low-level system inflammation—low-level insulin resistance | Low-rate inflammatory/immune cell migration into tissues Moderate-level organ damage— slow-rate repair processes (tissue homeostasis reversed) | Slightly higher IL-37 (0.8) TSH normal (1.9) |
| Low-level organ damage Low-level system inflammation—low-level insulin resistance | Moderate-to-rapid rate of inflammatory/immune cell migration into tissues Low-level organ damage—the need for repair processes to take place Increasing stimulus for IL-37 induction | Higher IL-37 (3.4) TSH normal (2.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majnarić, L.T.; Bosnić, Z.; Štefanić, M.; Wittlinger, T. Cross-Talk between the Cytokine IL-37 and Thyroid Hormones in Modulating Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions. Int. J. Mol. Sci. 2022, 23, 6456. https://doi.org/10.3390/ijms23126456
Majnarić LT, Bosnić Z, Štefanić M, Wittlinger T. Cross-Talk between the Cytokine IL-37 and Thyroid Hormones in Modulating Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions. International Journal of Molecular Sciences. 2022; 23(12):6456. https://doi.org/10.3390/ijms23126456
Chicago/Turabian StyleMajnarić, Ljiljana Trtica, Zvonimir Bosnić, Mario Štefanić, and Thomas Wittlinger. 2022. "Cross-Talk between the Cytokine IL-37 and Thyroid Hormones in Modulating Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions" International Journal of Molecular Sciences 23, no. 12: 6456. https://doi.org/10.3390/ijms23126456
APA StyleMajnarić, L. T., Bosnić, Z., Štefanić, M., & Wittlinger, T. (2022). Cross-Talk between the Cytokine IL-37 and Thyroid Hormones in Modulating Chronic Inflammation Associated with Target Organ Damage in Age-Related Metabolic and Vascular Conditions. International Journal of Molecular Sciences, 23(12), 6456. https://doi.org/10.3390/ijms23126456