
New Insights into the Determinants of Specificity in Human Type I Arginase: Generation of a Mutant That Is Only Active with Agmatine as Substrate

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
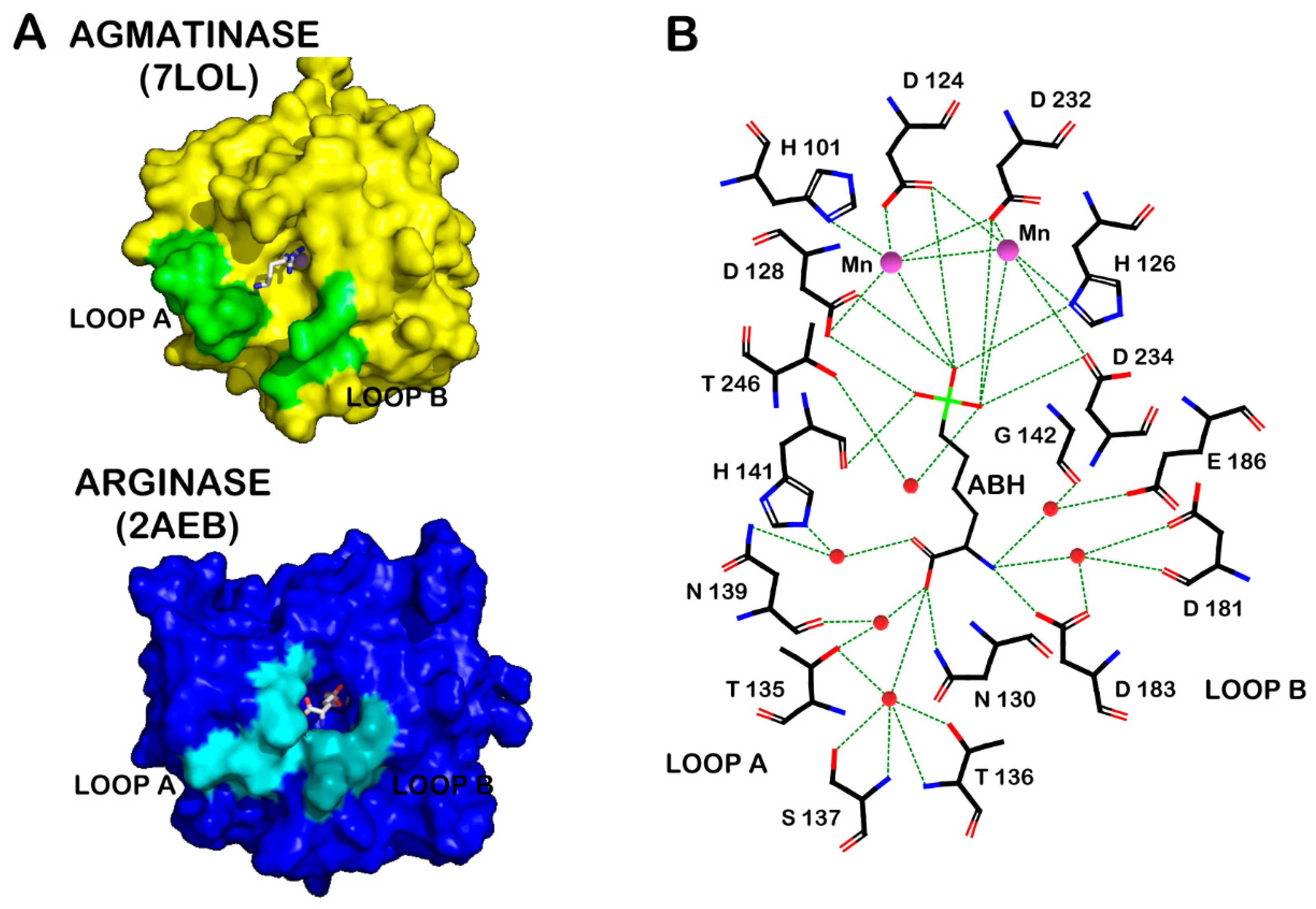
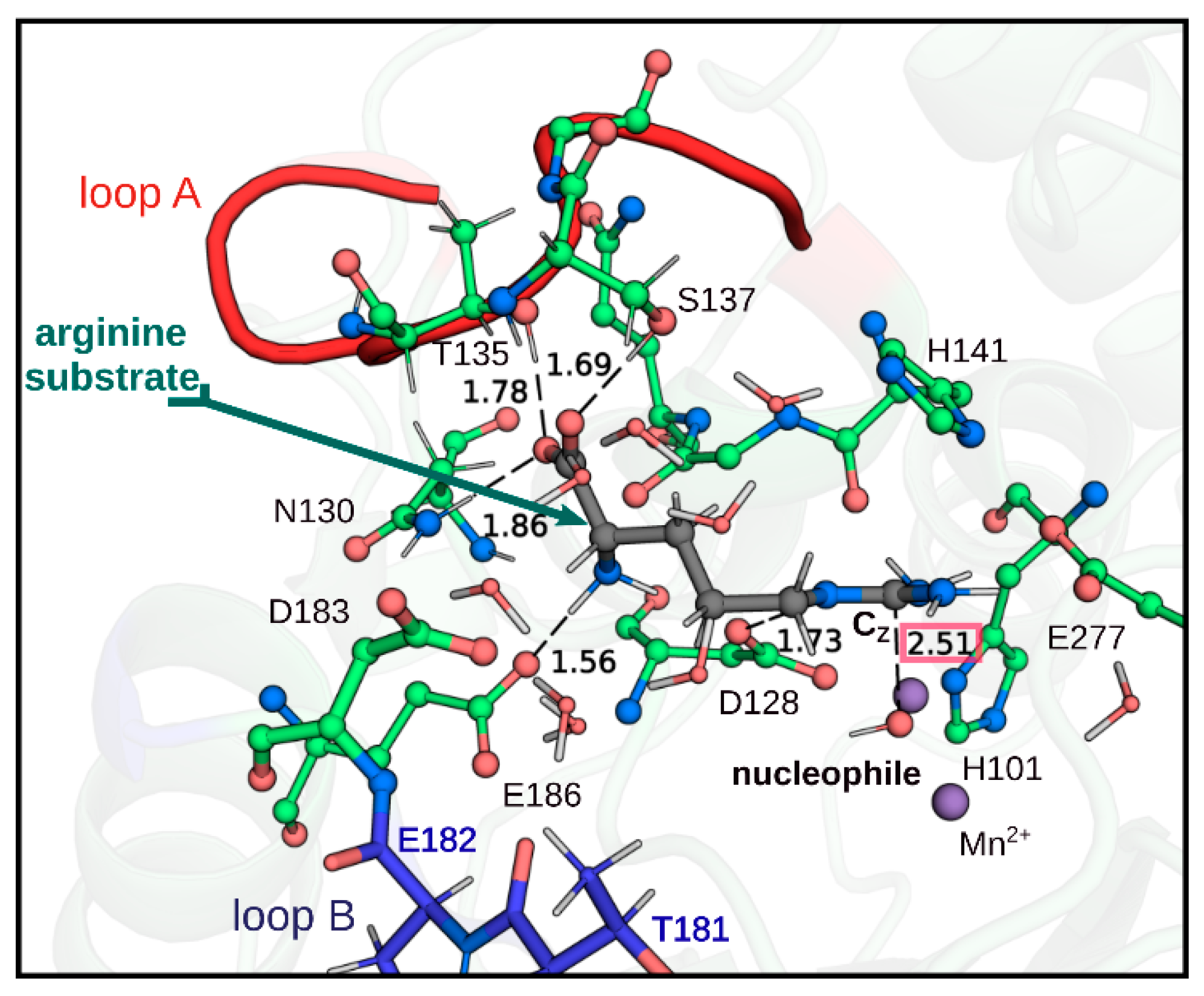
2.1. Loop A Mutagenesis in Human Arginase Type I
2.2. Loop B Mutagenesis in Human Arginase Type 1
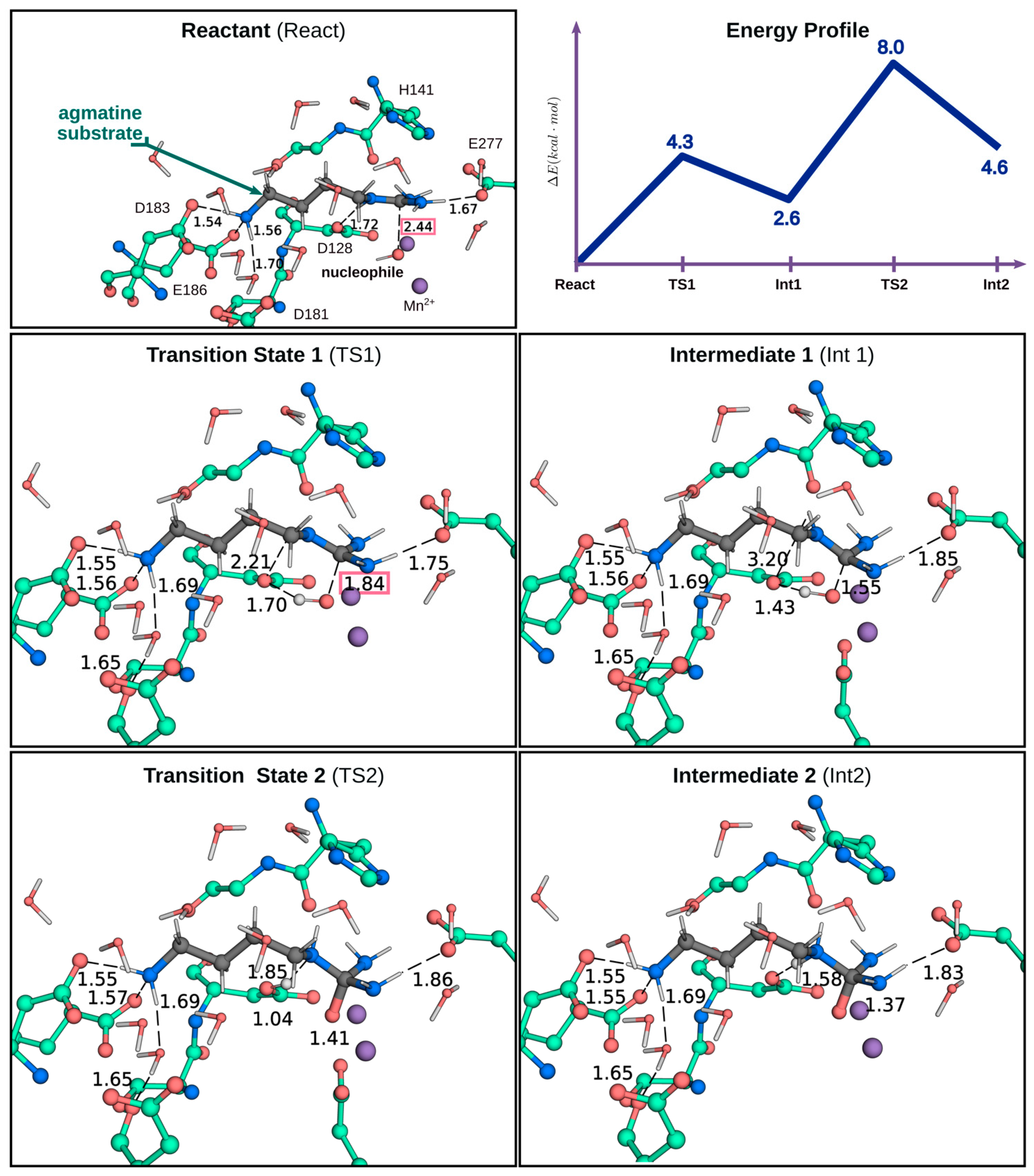
2.3. QM/MM Calculations to Estimate the Energy Barriers of the Hydrolysis Reaction in Chimera A2
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Enzyme Preparations
4.3. Site-Directed Mutagenesis
4.4. Enzymatic Assays and Kinetic Studies
4.5. Molecular Modeling and QM/MM Calculation of Arginase Enzyme with Mutation in A-Loop
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jenkinson, C.P.; Grody, W.W.; Cederbaum, S.D. Comparative properties of arginases. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1996, 114, 107–132. [Google Scholar] [CrossRef]
- Uribe, E.; Reyes, M.-B.; Martínez, I.; Mella, K.; Salas, M.; Tarifeño-Saldivia, E.; López, V.; García-Robles, M.; Martínez-Oyanedel, J.; Figueroa, M. Functional analysis of the Mn2+ requirement in the catalysis of ureohydrolases arginase and agmatinase—A historical perspective. J. Inorg. Biochem. 2020, 202, 110812. [Google Scholar] [CrossRef] [PubMed]
- Pudlo, M.; Demougeot, C.; Girard-Thernier, C. Arginase Inhibitors: A Rational Approach Over One Century: An exhaustive review of arginase inhibitors. Med. Res. Rev. 2017, 37, 475–513. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, N.; Cederbaum, S.D. Kinetics of inhibition of rat liver and kidney arginases by proline and branched-chain amino acids. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1986, 870, 181–184. [Google Scholar] [CrossRef]
- Cama, E.; Colleluori, D.; Emig, F.; Shin, H.; Kim, S.; Kim, N.; Traish, A.; Ash, D.; Christianson, D. Human Arginase II: Crystal Structure and Physiological Role in Male and Female Sexual Arousal. Biochemistry 2003, 42, 8445–8451. [Google Scholar] [CrossRef]
- Benítez, J.; García; Romero, N.; González, A.; Martínez-Oyanedel, J.; Figueroa, M.; Salas, M.; López, V.; García-Robles, M.; Dodd, P.R.; et al. Elena Uribe Metabolic strategies for the degradation of the neuromodulator agmatine in mammals. Metabolism 2018, 81, 35–44. [Google Scholar] [CrossRef]
- Cederbaum, S.D.; Yu, H.; Grody, W.W.; Kern, R.M.; Yoo, P.; Iyer, R.K. Arginases I and II: Do their functions overlap? Mol. Genet. Metab. 2004, 81, 38–44. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.; Zaagsma, J.; Meurs, H. The therapeutic potential of drugs targeting the arginase pathway in asthma. Expert Opin. Investig. Drugs 2005, 14, 1221–1231. [Google Scholar] [CrossRef]
- Kitowska, K.; Zakrzewicz, D.; Königshoff, M.; Chrobak, I.; Grimminger, F.; Seeger, W.; Bulau, P.; Eickelberg, O. Functional role and species-specific contribution of arginases in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L34–L45. [Google Scholar] [CrossRef]
- Maarsingh, H.; Zuidhof, A.; Bos, S.; van Duin, M.; Boucher, J.; Zaagsma, J.; Meurs, H. Arginase Inhibition Protects against Allergen-induced Airway Obstruction, Hyperresponsiveness, and Inflammation. Am. J. Respir. Crit. Care Med. 2008, 178, 565–573. [Google Scholar] [CrossRef]
- Esteban, S.; Clemente, C.; Koziol, A.; Gonzalo, P.; Rius, C.; Martínez, F.; Linares, P.; Chaparro, M.; Urzainqui, A.; Andrés, V.; et al. Endothelial MT1-MMP targeting limits intussusceptive angiogenesis and colitis via TSP1/nitric oxide axis. EMBO Mol. Med. 2020, 12, e10862. [Google Scholar] [CrossRef] [PubMed]
- Moretto, J.; Pudlo, M.; Demougeot, C. Human-based evidence for the therapeutic potential of arginase inhibitors in cardiovascular diseases. Drug Discov. Today 2021, 26, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Ouzounis, C.A.; Kyrpides, N.C. On the evolution of arginases and related enzymes. J. Mol. Evol. 1994, 39, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Perozich, J.; Hempel, J.; Morris, S.M. Roles of conserved residues in the arginase family. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1998, 1382, 23–37. [Google Scholar] [CrossRef]
- Sekowska, A.; Danchin, A.; Risler, J.-L. Phylogeny of related functions: The case of polyamine biosynthetic enzymes. Microbiology 2000, 146, 1815–1828. [Google Scholar] [CrossRef][Green Version]
- Schenk, S.; Mitić, N.; Gahan, L.; Ollis, D.; McGeary, P.; Guddat, L. Binuclear Metallohydrolases: Complex Mechanistic Strategies for a Simple Chemical Reaction. Acc. Chem. Res. 2012, 45, 1593–1603. [Google Scholar] [CrossRef]
- Alarcón, R.; Orellana, M.S.; Neira, B.; Uribe, E.; García, J.; Carvajal, N. Mutational analysis of substrate recognition by human arginase type I—Agmatinase activity of the N130D variant. FEBS J. 2006, 273, 5625–5631. [Google Scholar] [CrossRef]
- Lopez, V.; Alarcón, R.; Orellana, M.S.; Enríquez, P.; Uribe, E.; Martínez, J.; Carvajal, N. Insights into the interaction of human arginase II with substrate and manganese ions by site-directed mutagenesis and kinetic studies. Alteration of substrate specificity by replacement of Asn149 with Asp. FEBS J. 2005, 272, 4540–4548. [Google Scholar] [CrossRef]
- Shishova, E.Y.; Di Costanzo, L.; Emig, F.A.; Ash, D.E.; Christianson, D.W. Probing the Specificity Determinants of Amino Acid Recognition by Arginase. Biochemistry 2009, 48, 121–131. [Google Scholar] [CrossRef]
- Hyung, J.A.; Kyoung, H.K.; Lee, J.; Ha, J.Y.; Hyung, H.L.; Kim, D.; Yoon, H.J.; Kwon, A.R.; Se, W.S. Crystal structure of ag-matinase reveals structural conservation and inhibition mechanism of the ureohydrolase superfamily. J. Biol. Chem. 2004, 279, 50505–50513. [Google Scholar]
- Maturana, P.; Orellana, M.S.; Herrera, S.M.; Martínez, I.; Figueroa, M.; Martínez-Oyanedel, J.; Castro-Fernandez, V.; Uribe, E. Crystal Structure of Escherichia coli Agmatinase: Catalytic Mechanism and Residues Relevant for Substrate Specificity. Int. J. Mol. Sci. 2021, 22, 4769. [Google Scholar] [CrossRef] [PubMed]
- Reczkowski, R.S.; Ash, D.E. Rat Liver Arginase: Kinetic Mechanism, Alternate Substrates, and Inhibitors. Arch. Biochem. Biophys. 1994, 312, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Colleluori, D.M.; Reczkowski, R.S.; Emig, F.A.; Cama, E.; Cox, J.D.; Scolnick, L.R.; Compher, K.; Jude, K.; Han, S.; Viola, R.E.; et al. Probing the role of the hyper-reactive histidine residue of arginase. Arch. Biochem. Biophys. 2005, 444, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Hrabak, A.; Bajor, T.; Temesi, A. Comparison of Substrate and Inhibitor Specificity of Arginase and Nitricm Oxide (NO) Synthase for Arginine Analogs and Related Compounds in Murine and Rat Macrophages. Biochem. Biophys. Res. Commun. 1994, 198, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; Rodríguez, R.; López, N.; Uribe, E.; López, V.; Carvajal, N. Insights into the reaction mechanism of Escherichia coli agmatinase by site-directed mutagenesis and molecular modelling. Eur. J. Biochem. 2002, 269, 5522–5526. [Google Scholar] [CrossRef]
- Bewley, M.C.; Jeffrey, P.D.; Patchett, M.L.; Kanyo, Z.F.; Baker, E.N. Crystal structures of Bacillus caldovelox arginase in complex with substrate and inhibitors reveal new insights into activation, inhibition and catalysis in the arginase superfamily. Structure 1999, 7, 435–448. [Google Scholar] [CrossRef]
- Kanyo, Z.F.; Scolnick, L.R.; Ash, D.E.; Christianson, D.W. Structure of a unique binuclear manganese cluster in arginase. Nature 1996, 383, 554. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Sabio, G.; Mora, A.; Rodriguez, P.; Ochoa, A.; Centeno, F.; Christianson, D. Crystal structure of human arginase I at 1.29-Å resolution and exploration of inhibition in the immune response. Proc. Natl. Acad. Sci. USA 2005, 102, 13058–13063. [Google Scholar] [CrossRef]
- Grobben, Y.; Uitdehaag, J.; Willemsen-Seegers, N.; Tabak, W.; de Man, J.; Buijsman, R.; Zaman, G. Structural insights into human Arginase-1 pH dependence and its inhibition by the small molecule inhibitor CB-1158. J. Struct. Biol. X 2019, 4, 100014. [Google Scholar] [CrossRef]
- Kim, N.; Cox, D.; Baggio, R.; Emig, F.; Mistry, S.; Harper, S.; Speicher, D.; Morris, S., Jr.; Ash, D.; Traish, A.; et al. Probing Erectile Function: S -(2-Boronoethyl)-l-Cysteine Binds to Arginase as a Transition State Analogue and Enhances Smooth Muscle Relaxation in Human Penile Corpus Cavernosum. Biochemistry 2001, 40, 2678–2688. [Google Scholar] [CrossRef]
- Chitrakar, I.; Ahmed, S.F.; Torelli, A.T.; French, J.B. Structure of the E. coli agmatinase, SPEB. PLoS ONE 2021, 16, e0248991. [Google Scholar] [CrossRef] [PubMed]
- Chinard, F.P. Photometric estimation ofproline and omithíne. J. Biof. Chem. 1952, 199, 91–95. [Google Scholar] [CrossRef]
- Orellana, M.S.; López, V.; Uribe, E.; Fuentes, M.; Salas, M.; Carvajal, N. Insights into the interaction of human liver arginase with tightly and weakly bound manganese ions by chemical modification and site-directed mutagenesis studies. Arch. Biochem. Biophys. 2002, 403, 155–159. [Google Scholar] [CrossRef]
- Saito, T.; Kawakami, T.; Yamanaka, S.; Okumura, M. QM/MM study of hydrolysis of arginine catalysed by arginase. Mol. Phys. 2016, 114, 431–439. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Caballero, J.; Tuñón, I.; Hernández-Rodríguez, E.W.; Ruiz-Pernía, J.J. On the Nature of the Enzyme–Substrate Complex and the Reaction Mechanism in Human Arginase I. A Combined Molecular Dynamics and QM/MM Study. ACS Catal. 2020, 10, 8321–8333. [Google Scholar] [CrossRef]
- Carvajal, N.; Torres, C.; Uribe, E.; Salas, M. Interaction of arginase with metal ions: Studies of the enzyme from human liver and comparison with other arginases. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 1995, 112, 153–159. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Archibald, R.M. Colorimetric determination of urea. J. Biol. Chem. 1945, 157, 507–518. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Brooks, C.L., III; Brünger, A.; Karplus, M. Active site dynamics in protein molecules: A stochastic boundary molecular-dynamics approach. Biopolymers 1985, 24, 843–865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.; Grubmüller, H.; MacKerell, A. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Woodcock, H.; Hodošček, M.; Gilbert, A.; Gill, P.; Schaefer, H.; Brooks, B. Interfacing Q-Chem and CHARMM to perform QM/MM reaction path calculations. J. Comput. Chem. 2007, 28, 1485–1502. [Google Scholar] [CrossRef]
- Fischer, S.; Karplus, M. Conjugate peak refinement: An algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom. Chem. Phys. Lett. 1992, 194, 10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Arginine | Agmatine | ||||||
|---|---|---|---|---|---|---|---|
| Enzyme | kcat | Km | kcat/Km | Kiorn | kcat | Km | kcat/Km |
| (s−1) | (mM) | (M−1s−1) | (mM) | (s−1) | (mM) | (M−1s−1) | |
| N130D | 33 | 13.3 | 2.5 × 103 | 7.8 | 3.0 | 1.4 | 2.1 × 103 |
| S137C | 75 | 3.2 | 2.3 × 104 | 7.6 | 1.8 | 7.5 | 2.4 × 102 |
| N139D | 64 | 4.4 | 1.4 × 104 | 6.2 | 1.3 | 5.3 | 2.5 × 102 |
| N130D-S137C | 23 | 6.5 | 3.5 × 103 | 7.1 | 2.3 | 1.7 | 1.3 × 103 |
| N130D-N139D | 33 | 6.1 | 5.4 × 103 | 6.4 | 1.6 | 1.1 | 1.4 × 103 |
| S137C-N139D | 83 | 10.8 | 7.7 × 103 | 8.4 | 1.1 | 0.9 | 1.2 × 103 |
| N130D-S137C-N139D | 4 | 6.9 | 5.8 × 102 | 5.4 | 0.9 | 1.6 | 5.6 × 102 |
| WT-arginase | 190 | 1.5 | 1.3 × 105 | 1.0 | n/a | ||
| E. coli-agmatinase | n/a | 120 | 1.1 | 1.1 × 105 | |||
| Arginine | Agmatine | |||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | |
| WT-arginase | 1.5 ± 0.5 | 190 ± 10 | 1.3 × 105 | n/a | ||
| E. coli agmatinase | n/a | 1.1 ± 0.2 | 120 ± 10 | 1.1 × 105 | ||
| Chimera A1 I129T/N130Y/T131A | 2.5 ± 0.5 | 6.2 ± 0.4 | 2.4 × 103 | n/a | ||
| Chimera A2 I129T/N130Y/T131A+∆ P132-T134 | n/a | 6 ± 1 | 1.1 ± 0.2 | 1.8 × 102 | ||
| Chimera A3 I129T/N130Y/T131A ∆ P132–T134/N139F/L140D | n/a | n/a | ||||
| Chimera A4 I129T/N130Y/T131A/∆ P132–T134/T135N/T136G/S137C/G138E/N139F/L140D | n/a | n/a | ||||
| Chimera A5 (residues I129 t P144 according to Loop A in agmatinase) | n/a | n/a | ||||
| Substrate | Inhibitor | Inhibition Type | Kis (mM) | |
|---|---|---|---|---|
| WT-arginase | Arginine | Guanidine | Competitive | 56 ± 4 |
| Chimera A1 | Arginine |  | Competitive | 38 ± 6 |
| Chimera A2 | Agmatine | Competitive | 40 ± 6 | |
| WT-arginase | Arginine | Agmatine | Competitive | 42 ± 5 |
| Chimera A1 | Arginine |  | Competitive | 27 ± 2 |
| WT-arginase | Arginine | Ornithine | Competitive | 2 ± 0.5 |
| Chimera A1 | Arginine |  | Competitive | 60 ± 2 |
| Chimera A2 | Agmatine | Arginine | Competitive | 9 ± 3 |
 |
| Arginine | Agmatine | Ornithine Inhibition | Kis (mM) | Guanidine Inhibition | Kis (mM) | |||
|---|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (M−1 s−1) |  |  | ||||
| WT-arginase | 1.5 ± 0.5 | 190 ± 10 | 1.26 × 105 | n/a | Competitive | 2 ± 0.5 | Competitive | 60 ± 2 |
| D181T | 5.3 ± 0.8 | 191 ± 8 | 3.6 × 104 | n/a | Competitive | 1.6 ± 0.3 | Competitive | 85 ± 10 |
| V182E | 2.4 ± 0.2 | 187 ± 10 | 7.7 × 104 | n/a | Competitive | 3.2 ± 0.2 | Competitive | 89 ± 12 |
| D181T/V182E | 32 ± 5 | 188 ± 10 | 5.8 × 103 | n/a | Mixed | Kis = 71 ± 12 Kii = 203 ± 28 | Competitive | 80 ± 5 |
| D181T/V182E + ins F | 20 ± 5 | 8.1 ± 1 | 3.5 × 102 | n/a | Competitive | 18 ± 6 | Competitive | 70 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orellana, M.-S.; Jaña, G.A.; Figueroa, M.; Martínez-Oyanedel, J.; Medina, F.E.; Tarifeño-Saldivia, E.; Gatica, M.; García-Robles, M.Á.; Carvajal, N.; Uribe, E. New Insights into the Determinants of Specificity in Human Type I Arginase: Generation of a Mutant That Is Only Active with Agmatine as Substrate. Int. J. Mol. Sci. 2022, 23, 6438. https://doi.org/10.3390/ijms23126438
Orellana M-S, Jaña GA, Figueroa M, Martínez-Oyanedel J, Medina FE, Tarifeño-Saldivia E, Gatica M, García-Robles MÁ, Carvajal N, Uribe E. New Insights into the Determinants of Specificity in Human Type I Arginase: Generation of a Mutant That Is Only Active with Agmatine as Substrate. International Journal of Molecular Sciences. 2022; 23(12):6438. https://doi.org/10.3390/ijms23126438
Chicago/Turabian StyleOrellana, María-Soledad, Gonzalo A. Jaña, Maximiliano Figueroa, José Martínez-Oyanedel, Fabiola E. Medina, Estefanía Tarifeño-Saldivia, Marcell Gatica, María Ángeles García-Robles, Nelson Carvajal, and Elena Uribe. 2022. "New Insights into the Determinants of Specificity in Human Type I Arginase: Generation of a Mutant That Is Only Active with Agmatine as Substrate" International Journal of Molecular Sciences 23, no. 12: 6438. https://doi.org/10.3390/ijms23126438
APA StyleOrellana, M.-S., Jaña, G. A., Figueroa, M., Martínez-Oyanedel, J., Medina, F. E., Tarifeño-Saldivia, E., Gatica, M., García-Robles, M. Á., Carvajal, N., & Uribe, E. (2022). New Insights into the Determinants of Specificity in Human Type I Arginase: Generation of a Mutant That Is Only Active with Agmatine as Substrate. International Journal of Molecular Sciences, 23(12), 6438. https://doi.org/10.3390/ijms23126438