
Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome

 , , , and
, , , and 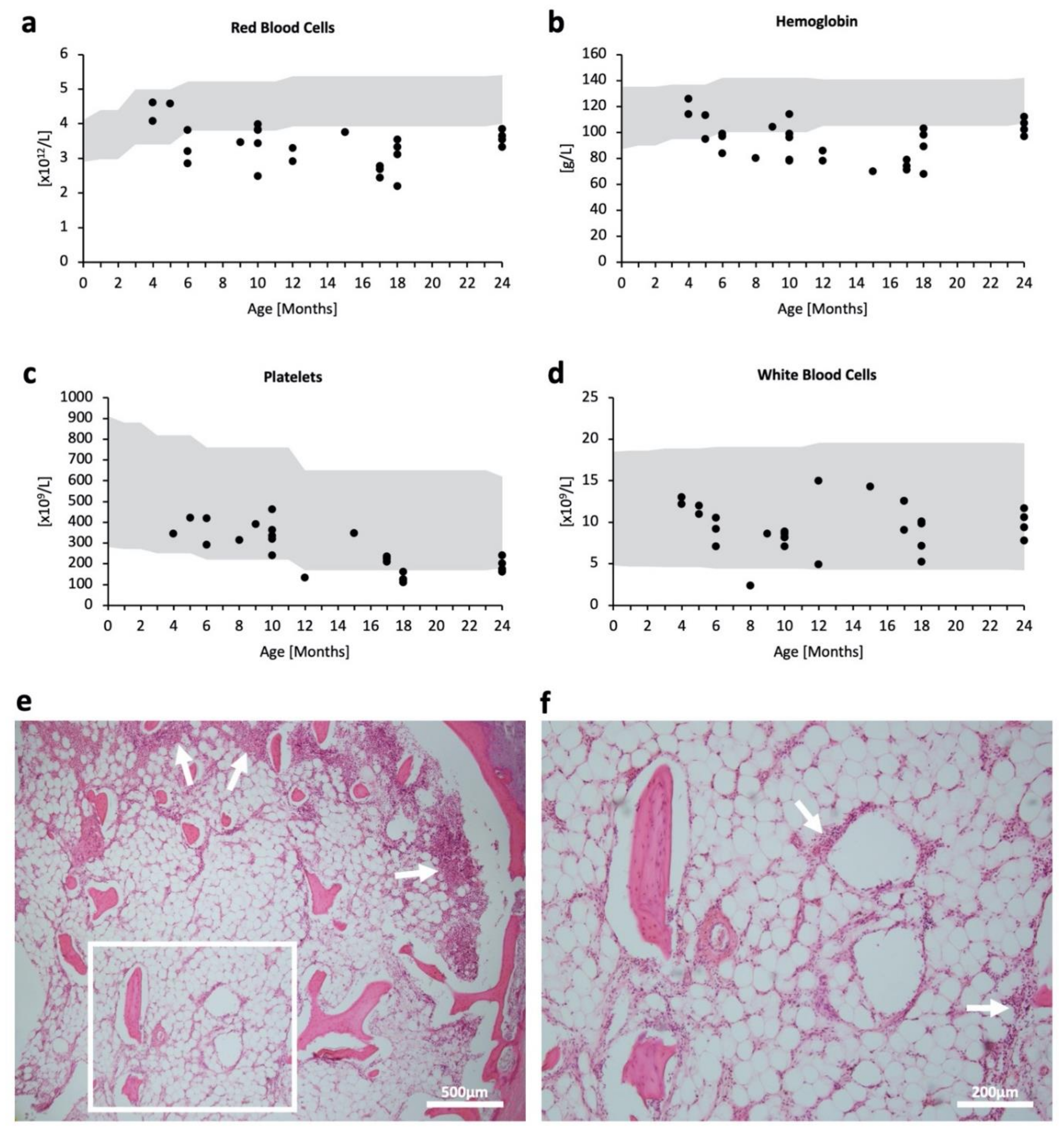{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
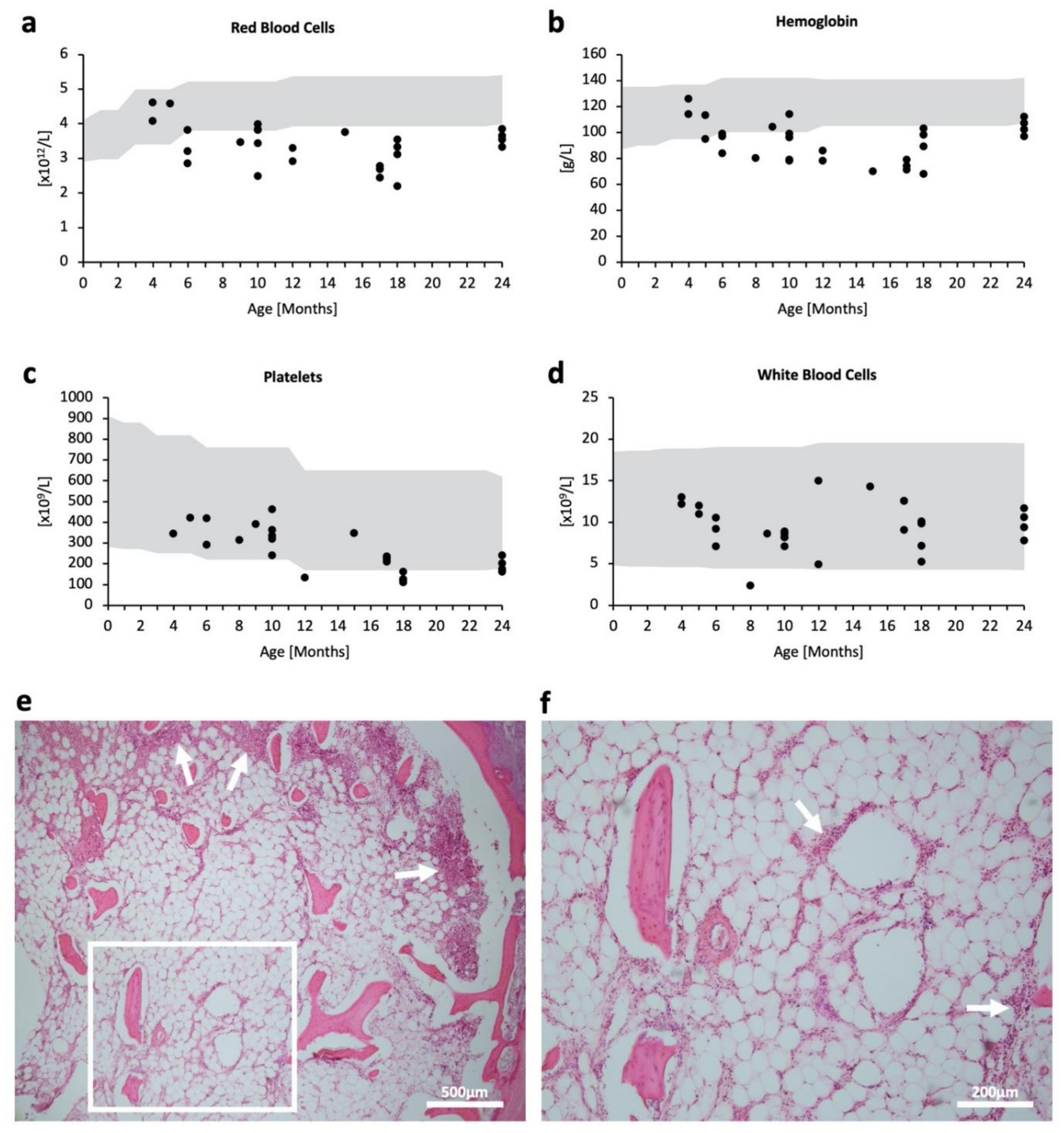
2.1. Hematopoietic Impairment in MPSPS
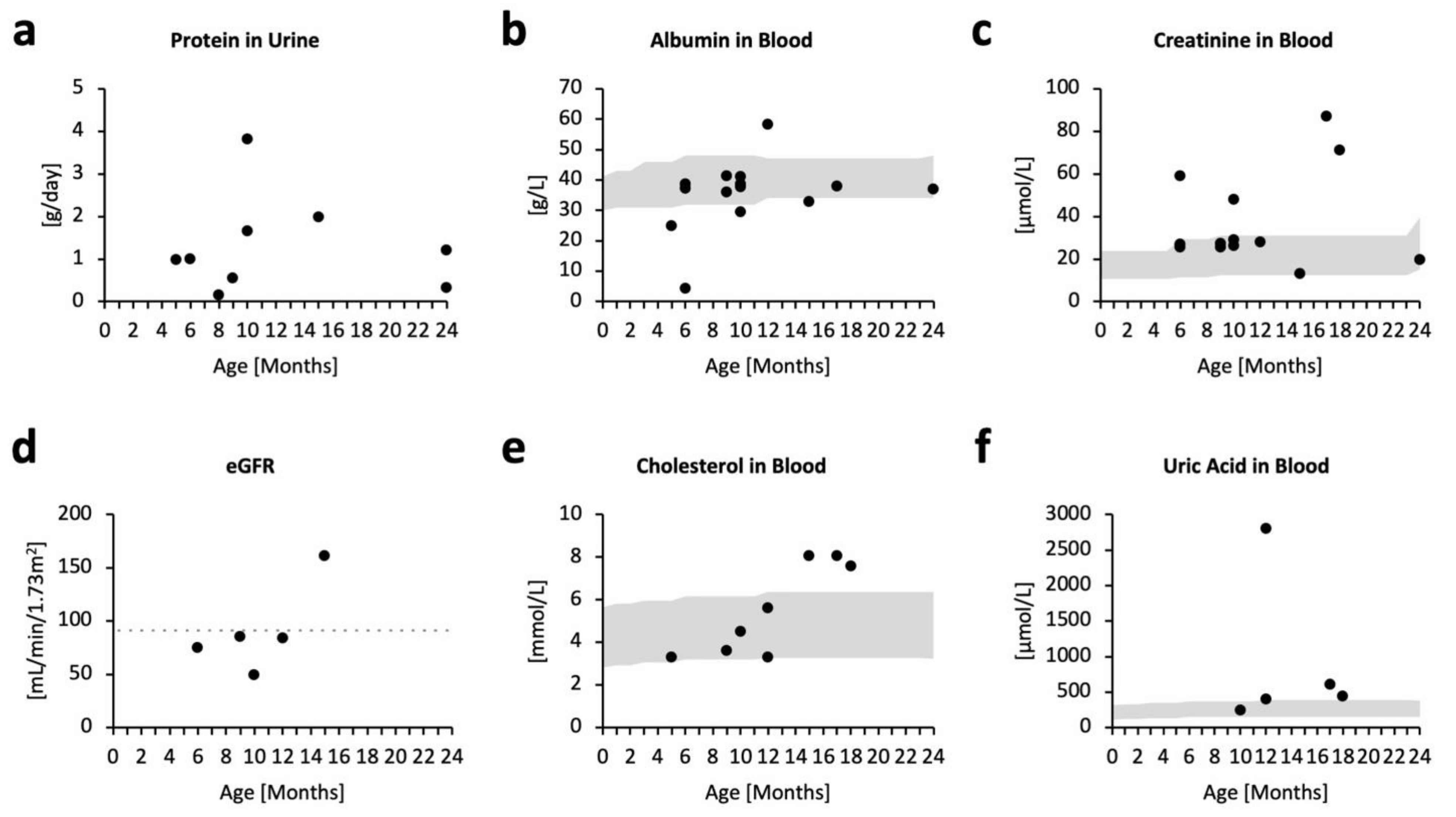
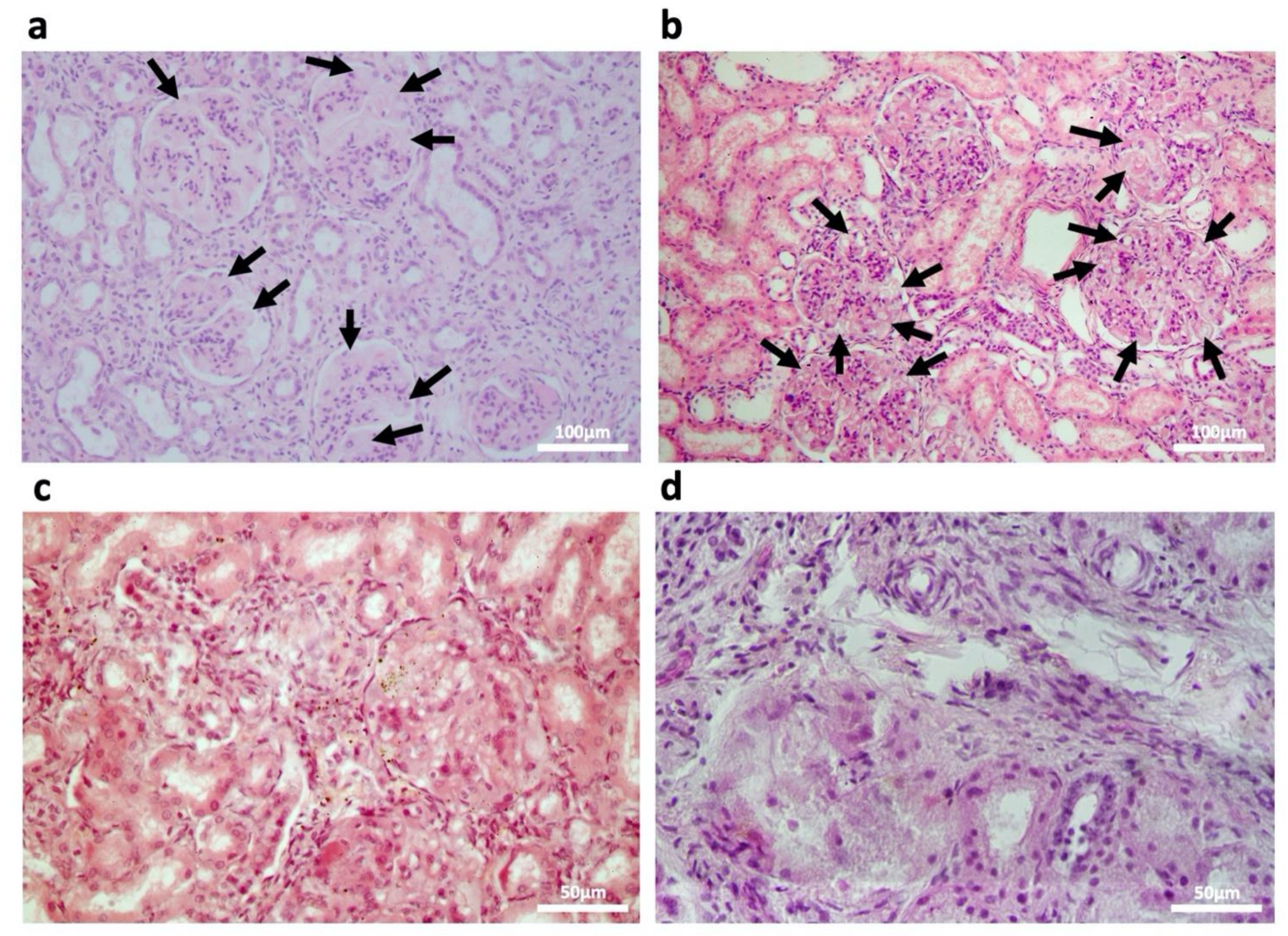
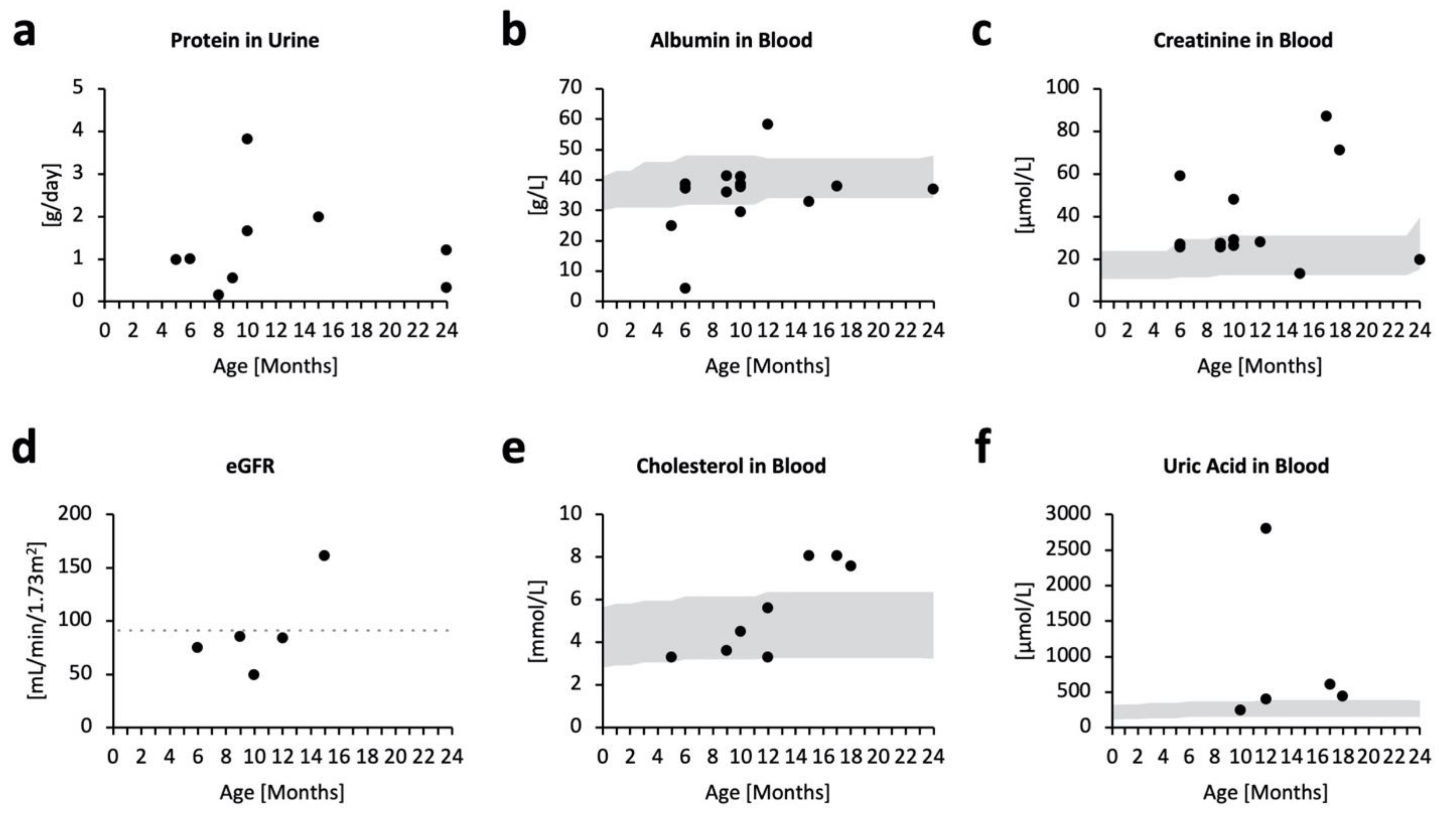
2.2. Kidney Impairment in MPSPS
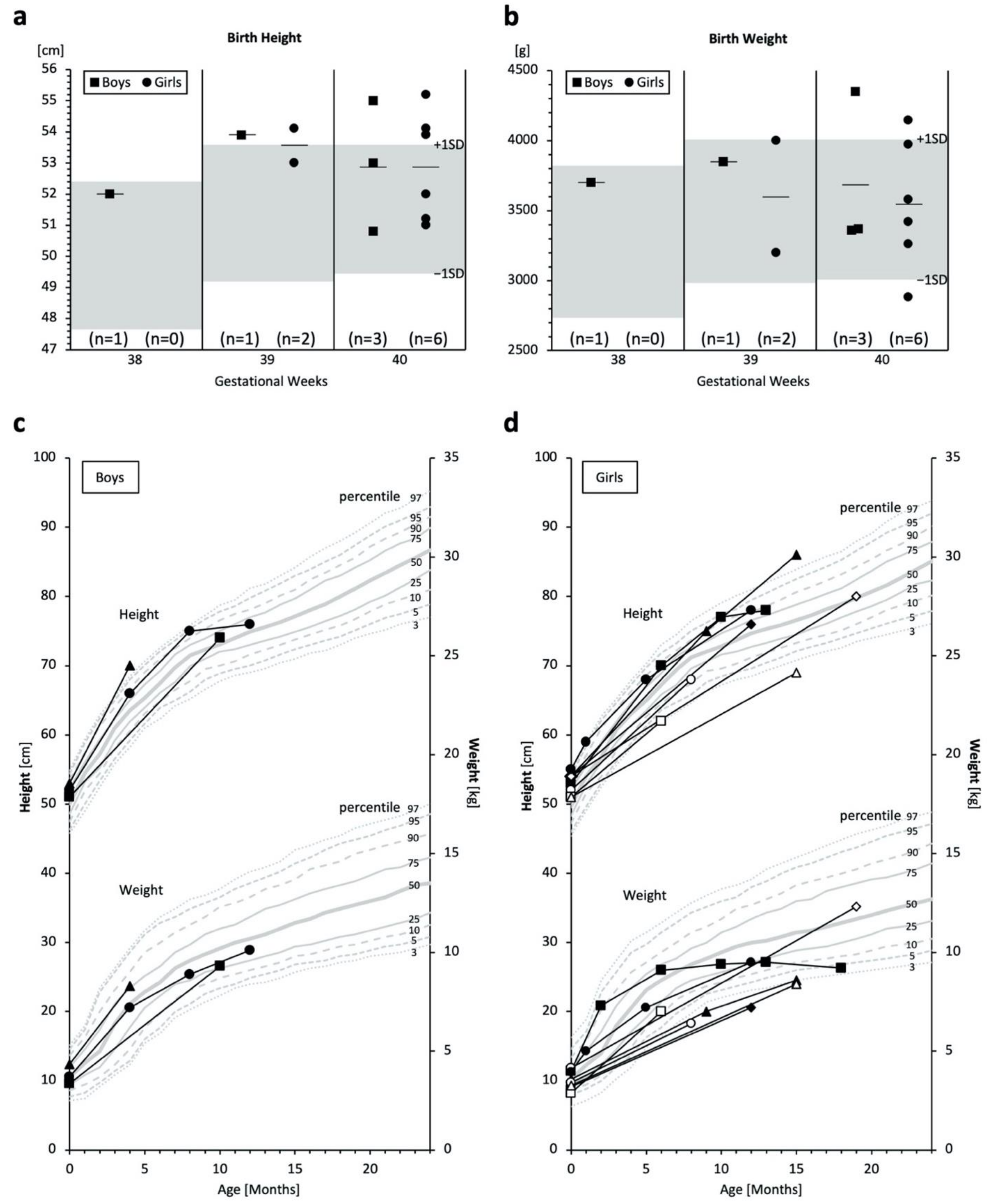
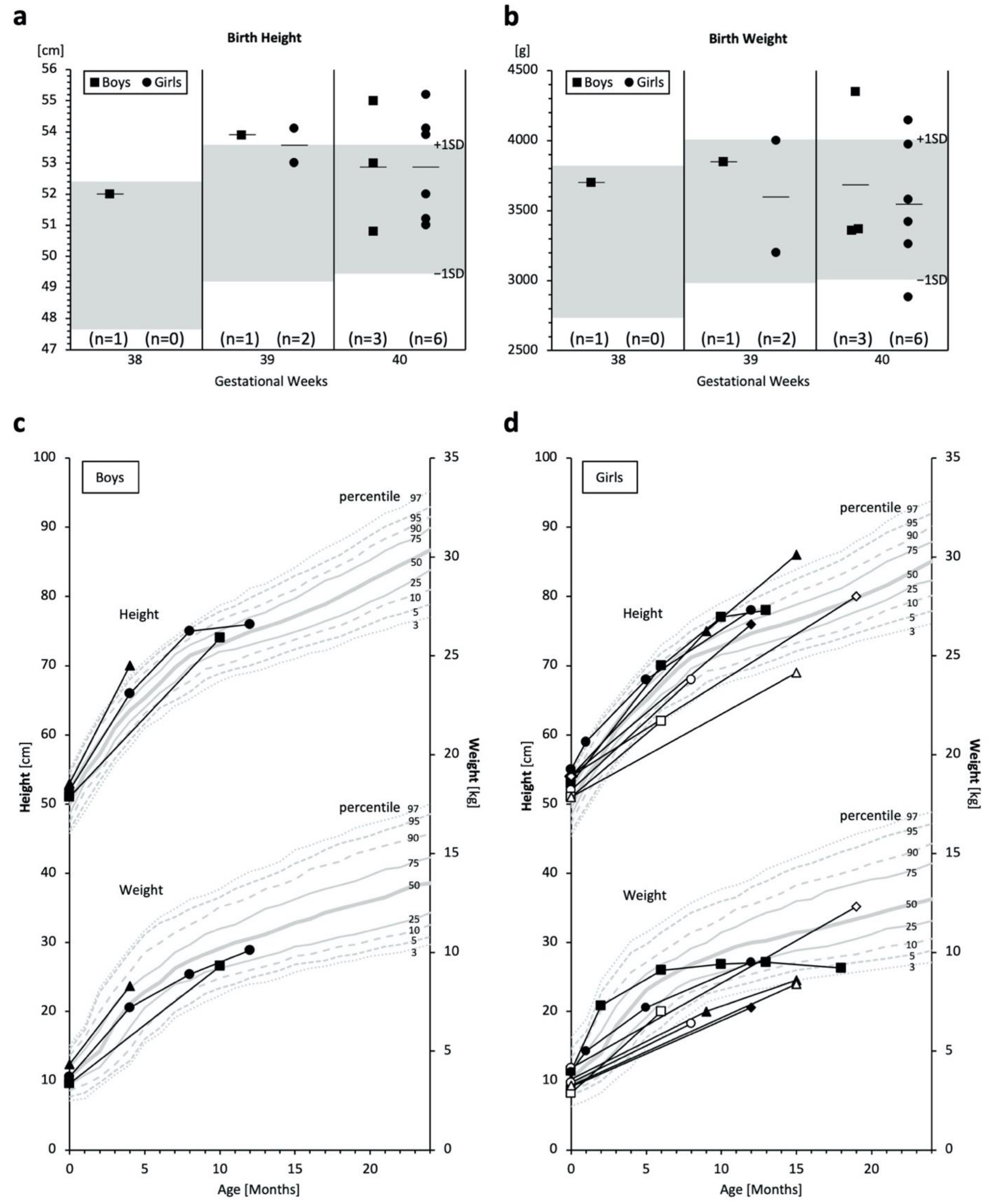
2.3. Birth Size and Growth
3. Discussion
4. Materials and Methods
4.1. Clinical Cases
4.2. Histological Analyses
4.3. Blood Count, Biochemical, and Physical Standard References
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Tomatsu, S.; Lavery, C.; Harmatz, P.; Scarpa, M.; Węgrzyn, G.; Orii, T. An Overview of Mucopolysaccharidoses: Diagnosis, Natural History, and Clinical Pictures. In Mucopolysaccharidoses Update (Metabolic Diseases—Laboratory and Clinical Research), 1st ed.; Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science: New York, NY, USA, 2018; Volume 1, pp. 17–19. ISBN 978-1-53613-986-0. [Google Scholar]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of mucopolysaccharidoses update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurinova, E.E.; Maksimova, N.R.; Sukhomyasova, A.L. Clinical Description of a Rare Autosomal Recessive Syndrome in the Yakut Children. Yakut Med. J. 2014, 2, 14–18. [Google Scholar]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A affects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 2017, 26, 173–183. [Google Scholar] [PubMed] [Green Version]
- Dursun, A.; Yalnizoglu, D.; Gerdan, O.F.; Yucel-Yilmaz, D.; Sagiroglu, M.S.; Yuksel, B.; Gucer, S.; Sivri, S.; Ozgul, R.K. A probable new syndrome with the storage disease phenotype caused by the VPS33A gene mutation. A probable new syndrome with the storage disease phenotype caused by the VPS33A gene mutation. Clin. Dysmorphol. 2017, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Faraguna, M.C.; Musto, F.; Crescitelli, V.; Iascone, M.; Spaccini, L.; Tonduti, D.; Fedeli, T.; Kullmann, G.; Canonico, F.; Cattoni, A.; et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes 2022, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Wartosch, L.; Günesdogan, U.; Graham, S.C.; Luzio, J.P. Recruitment of VPS33A to HOPS by VPS16 Is Required for Lysosome Fusion with Endosomes and Autophagosomes. Traffic 2015, 16, 727–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, E.V.; Shatunov, A.; Wartosch, L.; Moskvina, A.I.; Nikolaeva, L.E.; Bright, N.A.; Tylee, K.L.; Church, H.J.; Ballabio, A.; Luzio, J.P.; et al. The lysosomal disease caused by mutant VPS33A. Hum. Mol. Genet. 2019, 28, 2514–2530. [Google Scholar] [CrossRef] [PubMed]
- Atul Mehta, D.A.; Hughes, D.A. GeneReviews® [Internet] Fabry Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1292/ (accessed on 29 April 2022).
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C. Lysosome function in glomerular health and disease. Cell Tissue Res. 2021, 385, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, S. Electron microscopy illuminates the pathology of Fabry nephropathy. Nat. Rev. Nephrol. 2011, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Moore, M.; Lager, D. Fabry disease: A morphologic study of 11 cases. Mod. Pathol. 2006, 19, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Koga-Kobori, S.; Sawa, N.; Kido, R.; Sekine, A.; Mizuno, H.; Yamanouchi, M.; Hayami, N.; Suwabe, T.; Hoshino, J.; Kinowaki, K.; et al. Fabry Disease on Peritoneal Dialysis with Cardiac Involvement. Intern. Med. 2021, 60, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Takahashi-Fujigasaki, J.; Fukuda, T.; Sakurai, K.; Shimada, Y.; Nomura, K.; Ariga, M.; Ohashi, T.; Eto, Y.; Otomo, T.; et al. Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol. Genet. Metab. 2011, 102, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T. Pediatric Clinical Test Standard Values. In Higher-Level Pediatric Clinical Tests: How to Choose and Think Based on Pathophysiology; Ozono, K., Ed.; Sougou Igaku: Tokyo, Japan, 2013; Volume 3. [Google Scholar]
- Nagai, T.; Uemura, O.; Honda, M.; Matsuyama, K.; Akioka, Y.; Awazu, M.; Iijima, K.; Ikezumi, Y.; Ishikura, K.; Ito, S.; et al. GFR estimation formula for Japanese children (2–12 years old) -Interim report-. J. Japan. Soc. Ped. Kid. Dis. 2010, 23, 245–249. [Google Scholar]
- Chasnyk, V.G.; Burtseva, T.E.; Dranaeva, G.G. Dynamics of Anthropometric Characteristics and Blood Pressure in Children of Republic of Sakha (Yakutia); Izdatelstvo Sphera: Moscow, Russia, 2017; pp. 56–157. [Google Scholar]
- Dementeva, G.M.; Korotkaya, E.V. Differential assessment of low-birth-weight children. Vopr. Ohr. Materin. I Det. 1981, 2, 15–20. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sofronova, V.; Iwata, R.; Moriya, T.; Loskutova, K.; Gurinova, E.; Chernova, M.; Timofeeva, A.; Shvedova, A.; Vasilev, F.; Novgorodova, S.; et al. Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2022, 23, 5851. https://doi.org/10.3390/ijms23105851
Sofronova V, Iwata R, Moriya T, Loskutova K, Gurinova E, Chernova M, Timofeeva A, Shvedova A, Vasilev F, Novgorodova S, et al. Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome. International Journal of Molecular Sciences. 2022; 23(10):5851. https://doi.org/10.3390/ijms23105851
Chicago/Turabian StyleSofronova, Viktoriia, Rina Iwata, Takuya Moriya, Kiunniai Loskutova, Elizaveta Gurinova, Mairanush Chernova, Anastasia Timofeeva, Anna Shvedova, Filipp Vasilev, Saina Novgorodova, and et al. 2022. "Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome" International Journal of Molecular Sciences 23, no. 10: 5851. https://doi.org/10.3390/ijms23105851
APA StyleSofronova, V., Iwata, R., Moriya, T., Loskutova, K., Gurinova, E., Chernova, M., Timofeeva, A., Shvedova, A., Vasilev, F., Novgorodova, S., Terawaki, S., Moriwaki, T., Sukhomyasova, A., Maksimova, N., & Otomo, T. (2022). Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome. International Journal of Molecular Sciences, 23(10), 5851. https://doi.org/10.3390/ijms23105851