
Role of miRNAs in Human T Cell Leukemia Virus Type 1 Induced T Cell Leukemia: A Literature Review and Bioinformatics Approach

 , , ,
, , ,  and
and 
Abstract
1. Introduction
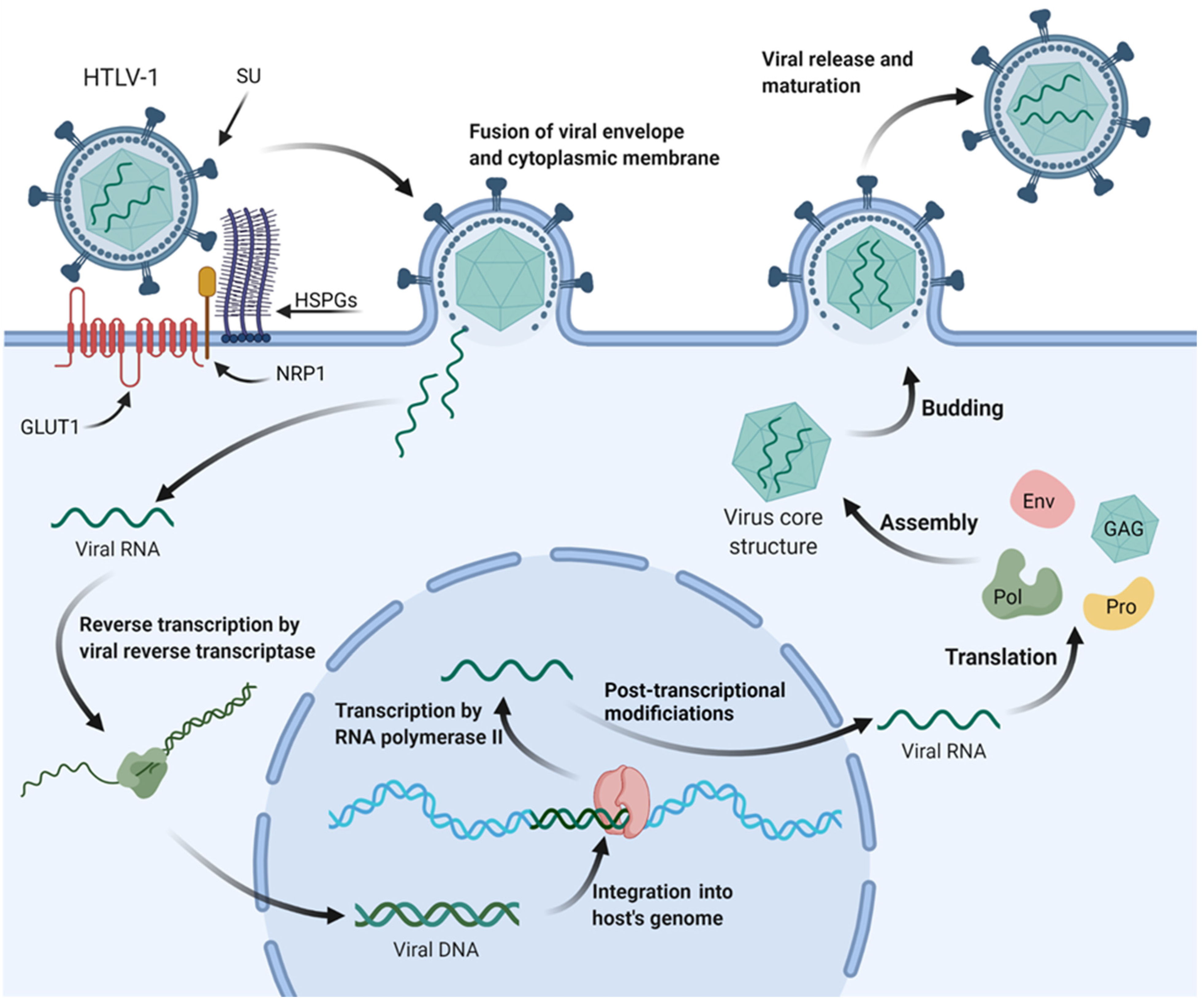
1.1. HTLV-1 Viral Structure and Infection Mechanisms
1.2. Leukemogenesis Pathways to Adult T Cell Leukemia
1.3. MicroRNAs
2. Literature Review and Bioinformatic Analysis Approach
3. TP53 Mutations and Modulation in HTLV-1 Infection
4. Pathways Identified as Deregulated in ATL
4.1. WNT Canonical and Non-Canonical Pathways
4.2. TGF-β Dual Roles in Cancer
4.3. RAS and MAPK Signaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tagaya, Y.; Gallo, R.C. The Exceptional Oncogenicity of HTLV-1. Front. Microbiol. 2017, 8, 1425. [Google Scholar] [CrossRef]
- Saito, M.; Matsuzaki, T.; Satou, Y.; Yasunaga, J.I.; Saito, K.; Arimura, K.; Matsuoka, M.; Ohara, Y. In Vivo Expression of the HBZ Gene of HTLV-1 Correlates with Proviral Load, Inflammatory Markers and Disease Severity in HTLV-1 Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP). Retrovirology 2009, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Rosadas, C.; Menezes, M.L.B.; Galvão-Castro, B.; Assone, T.; Miranda, A.E.; Aragón, M.G.; Caterino-De-araujo, A.; Taylor, G.P.; Ishak, R. Blocking HTLV-1/2 Silent Transmission in Brazil: Current Public Health Policies and Proposal for Additional Strategies. PLoS Negl. Trop. Dis. 2021, 15, e0009717. [Google Scholar] [CrossRef] [PubMed]
- Vieira, B.A.; Bidinotto, A.B.; Dartora, W.J.; Pedrotti, L.G.; de Oliveira, V.M.; Wendland, E.M. Prevalence of Human T-Lymphotropic Virus Type 1 and 2 (HTLV-1/-2) Infection in Pregnant Women in Brazil: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 15367. [Google Scholar] [CrossRef]
- Garcia, I.F.d.S.; Hennington, É.A. HTLV: A Stigmatizing Infection? Cad. Saude Publica 2019, 35, e00005419. [Google Scholar] [CrossRef]
- Oliveira-Filho, A.B.; Araújo, A.P.S.; Souza, A.P.C.; Gomes, C.M.; Silva-Oliveira, G.C.; Martins, L.C.; Fischer, B.; Machado, L.F.A.; Vallinoto, A.C.R.; Ishak, R.; et al. Human T-Lymphotropic Virus 1 and 2 among People Who Used Illicit Drugs in the State of Pará, Northern Brazil. Sci. Rep. 2019, 9, 14750. [Google Scholar] [CrossRef]
- Forlani, G.; Shallak, M.; Accolla, R.S.; Romanelli, M.G. HTLV-1 Infection and Pathogenesis: New Insights from Cellular and Animal Models. Int. J. Mol. Sci. 2021, 22, 8001. [Google Scholar] [CrossRef]
- Franchini, G.; Ambinder, R.F.; Barry, M. Viral Disease in Hematology. Hematology 2000, 2000, 409–423. [Google Scholar] [CrossRef]
- Delamarre, L.; Rosenberg, A.R.; Pique, C.; Pham, D.; Callebaut, I.; Dokhélar, M.C. The HTLV-I Envelope Glycoproteins: Structure and Functions. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13, S85–S91. [Google Scholar] [CrossRef]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.P.; Al-Saleem, J.; Green, P.L. Comparative Virology of HTLV-1 and HTLV-2. Retrovirology 2019, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Eusebio-Ponce, E.; Anguita, E.; Paulino-Ramirez, R.; Candel, F.J. HTLV-1 Infection: An Emerging Risk. Pathogenesis, Epidemiology, Diagnosis and Associated Diseases. Rev. Española Quimioter. 2019, 32, 485. [Google Scholar]
- Manns, A.; Hisada, M.; La Grenade, L. Human T-Lymphotropic Virus Type I Infection. Lancet Lond. Engl. 1999, 353, 1951–1958. [Google Scholar] [CrossRef]
- Brites, C.; Grassi, M.F.; Quaresma, J.A.S.; Ishak, R.; Vallinoto, A.C.R. Pathogenesis of HTLV-1 Infection and Progression Biomarkers: An Overview. Braz. J. Infect. Dis. 2021, 25, 101594. [Google Scholar] [CrossRef]
- Melamed, A.; Laydon, D.J.; Gillet, N.A.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R.M. Genome-Wide Determinants of Proviral Targeting, Clonal Abundance and Expression in Natural HTLV-1 Infection. PLOS Pathog. 2013, 9, e1003271. [Google Scholar] [CrossRef]
- Wattel, E.; Cavrois, M.; Gessain, A.; Wain-Hobson, S. Clonal Expansion of Infected Cells: A Way of Life for HTLV-I. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13, S92–S99. [Google Scholar] [CrossRef]
- Roucoux, D.F.; Wang, B.; Smith, D.; Nass, C.C.; Smith, J.; Hutching, S.T.; Newman, B.; Lee, T.-H.; Chafets, D.M.; Murphy, E.L. A Prospective Study of Sexual Transmission of Human T Lymphotropic Virus (HTLV)–I and HTLV-II. J. Infect. Dis. 2005, 191, 1490–1497. [Google Scholar] [CrossRef]
- Wu, K.; Bottazzi, M.E.; De La Fuente, C.; Deng, L.; Gitlin, S.D.; Maddukuri, A.; Dadgar, S.; Li, H.; Vertes, A.; Pumfery, A.; et al. Protein Profile of Tax-Associated Complexes. J. Biol. Chem. 2004, 279, 495–508. [Google Scholar] [CrossRef]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I between Lymphocytes by Virus-Induced Polarization of the Cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef]
- Gross, C.; Thoma-Kress, A.K. Molecular Mechanisms of HTLV-1 Cell-to-Cell Transmission. Viruses 2016, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N. B′-Protein Phosphatase 2A Is a Functional Binding Partner of Delta-Retroviral Integrase. Nucleic Acids Res. 2016, 44, 364. [Google Scholar] [CrossRef] [PubMed]
- Tasayco-Magallanes, E.; Miranda-Ulloa, E.; Romero-Ruiz, S.; Cárdenas-Bustamante, F.; Briceño-Espinoza, R.; Yana-Calatayud, B. Evaluation of Two Brands of Test of ELISA for the Diagnosis of HTLV-1 against Peruvian Samples. Rev. Chil. Infectol. 2020, 37, 780–783. [Google Scholar] [CrossRef]
- Cook, L.; Melamed, A.; Yaguchi, H.; Bangham, C.R. The Impact of HTLV-1 on the Cellular Genome. Curr. Opin. Virol. 2017, 26, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Giam, C.Z.; Semmes, O.J. HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma—A Tale of Two Proteins: Tax and HBZ. Viruses 2016, 8, 161. [Google Scholar] [CrossRef]
- Ernzen, K.J.; Panfil, A.R. Regulation of HTLV-1 Transformation. Biosci. Rep. 2022, 42, BSR20211921. [Google Scholar] [CrossRef]
- Pereira, W.A.; Mesquita, E.M. Vírus Linfotrópico de Células T Humana (HTLV): Doenças Associadas e Dificuldades No Diagnóstico e Tratamento. Rev. Investig. Bioméd. 2016, 8, 92–101. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human T-Cell Leukaemia Virus Type 1 (HTLV-1) Infectivity and Cellular Transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- Majorovits, E.; Nejmeddine, M.; Tanaka, Y.; Taylor, G.P.; Fuller, S.D.; Bangham, C.R.M. Human T-Lymphotropic Virus-1 Visualized at the Virological Synapse by Electron Tomography. PLoS ONE 2008, 3, e2251. [Google Scholar] [CrossRef]
- Yoshida, N.; Weinstock, D.M. Lymphoid Neoplasia: Clinicogenetic Risk Modeling in ATL. Blood 2018, 131, 159. [Google Scholar] [CrossRef]
- Shimoyama, M. Diagnostic Criteria and Classification of Clinical Subtypes of Adult T-Cell Leukaemia-Lymphoma. A Report from the Lymphoma Study Group (1984-87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Thorstensson, R.; Albert, J.; Andersson, S. Strategies for Diagnosis of HTLV-I and -II. Transfusion 2002, 42, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Brand, H.; Alves, J.G.B.; Pedrosa, F.; Lucena-Silva, N. Leucemia de Células T Do Adulto. Rev. Bras. Hematol. Hemoter. 2009, 31, 375–383. [Google Scholar] [CrossRef][Green Version]
- Franchini, G. HTLV-1 and HIV-1 “Accessory” Proteins: A Misleading Misnomer. Mol. Aspects Med. 2010, 31, 331–332. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.M.; Zanovello, P.; Watanabe, T.; Ciminale, V. The Metwork in Normal- and HTLV-1-Transformed T Cells. Adv. Cancer Res. 2012, 113, 45–83. [Google Scholar] [CrossRef] [PubMed]
- Simonson, B.; Das, S. MicroRNA Therapeutics: The Next Magic Bullet? Mini Rev. Med. Chem. 2015, 15, 467. [Google Scholar] [CrossRef]
- Bittencourt, A.L.; Farré, L. Leucemia/Linfoma de Células T Do Adulto. An. Bras. Dermatol. 2008, 83, 351–359. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef]
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of MicroRNA Biogenesis and Its Crosstalk with Other Cellular Pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying MiRNAs, Targets and Functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in Cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Moles, R.; Nicot, C. The Emerging Role of MiRNAs in HTLV-1 Infection and ATLL Pathogenesis. Viruses 2015, 7, 4047. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.; Valadão de Souza, D.R.; Pessôa, R.; Pietrobon, A.J.; Nukui, Y.; Pereira, J.; Casseb, J.; Penalva de Oliveira, A.C.; Loureiro, P.; da Silva Duarte, A.J.; et al. Global Expression of Noncoding RNome Reveals Dysregulation of Small RNAs in Patients with HTLV-1–Associated Adult T-Cell Leukemia: A Pilot Study. Infect. Agent. Cancer 2021, 16, 4. [Google Scholar] [CrossRef]
- Fayyad-Kazan, M.; ElDirani, R.; Hamade, E.; El Majzoub, R.; Akl, H.; Bitar, N.; Fayyad-Kazan, H.; Badran, B. Circulating MiR-29c, MiR-30c, MiR-193a-5p and MiR-885-5p: Novel Potential Biomarkers for HTLV-1 Infection Diagnosis. Infect. Genet. Evol. 2019, 74, 103938. [Google Scholar] [CrossRef]
- Sharma, V.K.; Raimondi, V.; Ruggero, K.; Pise-Masison, C.A.; Cavallari, I.; Silic-Benussi, M.; Ciminale, V.; D’Agostino, D.M. Expression of MiR-34a in T-Cells Infected by Human T-Lymphotropic Virus 1. Front. Microbiol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Moles, R.; Bellon, M.; Nicot, C. STAT1: A Novel Target of MiR-150 and MiR-223 Is Involved in the Proliferation of HTLV-I–Transformed and ATL Cells. Neoplasia 2015, 17, 449–462. [Google Scholar] [CrossRef]
- Vernin, C.; Thenoz, M.; Pinatel, C.; Gessain, A.; Gout, O.; Delfau-Larue, M.H.; Nazaret, N.; Legras-Lachuer, C.; Wattel, E.; Mortreux, F. HTLV-1 BZIP Factor HBZ Promotes Cell Proliferation and Genetic Instability by Activating OncomiRs. Cancer Res. 2014, 74, 6082–6093. [Google Scholar] [CrossRef]
- Ruggero, K.; Guffanti, A.; Corradin, A.; Sharma, V.K.; De Bellis, G.; Corti, G.; Grassi, A.; Zanovello, P.; Bronte, V.; Ciminale, V.; et al. Small Noncoding RNAs in Cells Transformed by Human T-Cell Leukemia Virus Type 1: A Role for a TRNA Fragment as a Primer for Reverse Transcriptase. J. Virol. 2014, 88, 3612. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M. Important Roles of Cellular MicroRNA MiR-155 in Leukemogenesis by Human T-Cell Leukemia Virus Type 1 Infection. ISRN Microbiol. 2012, 2012, 978607. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Quann, K.; Pandya, D.; Singh, S.; Khan, Z.K.; Jain, P. HTLV-1 Tax Mediated Downregulation of MiRNAs Associated with Chromatin Remodeling Factors in T Cells with Stably Integrated Viral Promoter. PLoS ONE 2012, 7, e34490. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Nakano, K.; Miyake, A.; Yamochi, T.; Kagami, Y.; Tsutsumi, A.; Matsuda, Y.; Sato-Otsubo, A.; Muto, S.; Utsunomiya, A.; et al. Polycomb-Mediated Loss of MiR-31 Activates NIK-Dependent NF-ΚB Pathway in Adult T Cell Leukemia and Other Cancers. Cancer Cell 2012, 21, 121–135. [Google Scholar] [CrossRef]
- Tomita, M.; Tanaka, Y.; Mori, N. MicroRNA MiR-146a Is Induced by HTLV-1 Tax and Increases the Growth of HTLV-1-Infected T-Cells. Int. J. Cancer 2012, 130, 2300–2309. [Google Scholar] [CrossRef]
- Bellon, M.; Lepelletier, Y.; Hermine, O.; Nicot, C. Deregulation of MicroRNA Involved in Hematopoiesis and the Immune Response in HTLV-I Adult T-Cell Leukemia. Blood 2009, 113, 4914–4917. [Google Scholar] [CrossRef]
- Pichler, K.; Schneider, G.; Grassmann, R. MicroRNA MiR-146a and Further Oncogenesis-Related Cellular MicroRNAs Are Dysregulated in HTLV-1-Transformed T Lymphocytes. Retrovirology 2008, 5, 100. [Google Scholar] [CrossRef]
- Yeung, M.L.; Yasunaga, J.I.; Bennasser, Y.; Dusetti, N.; Harris, D.; Ahmad, N.; Matsuoka, M.; Jeang, K.T. Roles for MicroRNAs, MiR-93 and MiR-130b, and TP53INP1 Tumor Suppressor in Cell Growth Dysregulation by HTLV-1. Cancer Res. 2008, 68, 8976. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting Effective MicroRNA Target Sites in Mammalian MRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. MiRDB: An Online Database for Prediction of Functional MicroRNA Targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Hu, H. TarPmiR: A New Approach for MicroRNA Target Site Prediction. Bioinformatics 2016, 32, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, Y.C.D.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA-Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New Approach for Understanding Genome Variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. MiRWalk: An Online Resource for Prediction of MicroRNA Binding Sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. P53, Cancer and the Immune Response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef]
- Borrero, L.J.H.; El-Deiry, W.S. Tumor Suppressor P53: Biology, Signaling Pathways, and Therapeutic Targeting. Biochim. Biophys. Acta. Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef]
- Yu, D.; Xu, Z.; Cheng, X.; Qin, J. The Role of MiRNAs in MDMX-P53 Interplay. J. Evid. Based Med. 2021, 14, 152–160. [Google Scholar] [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Wolff, R.K.; Sakoda, L.C.; Samowitz, W.S.; Herrick, J.S. The P53-Signaling Pathway and Colorectal Cancer: Interactions between Downstream P53 Target Genes and MiRNAs. Genomics 2019, 111, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Fochi, S.; Mutascio, S.; Bertazzoni, U.; Zipeto, D.; Romanelli, M.G. HTLV Deregulation of the NF-ΚB Pathway: An Update on Tax and Antisense Proteins Role. Front. Microbiol. 2018, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Ducasa, N.; Grasso, D.; Benencio, P.; Papademetrio, D.L.; Biglione, M.; Kashanchi, F.; Berini, C.; Garcia, M.N. Autophagy in Human T-Cell Leukemia Virus Type 1 (HTLV-1) Induced Leukemia. Front. Oncol. 2021, 11, 967. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant P53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Ishida, T.; Masaki, A.; Murase, T.; Takeshita, M.; Muto, R.; Iwasaki, H.; Ito, A.; Kusumoto, S.; Nakano, N.; et al. Clinical Significance of TP53 Mutations in Adult T-Cell Leukemia/Lymphoma. Br. J. Haematol. 2021, 195, 571–584. [Google Scholar] [CrossRef]
- Cook, L.B.M. Adult T-Cell Leukaemia/Lymphoma and the Trouble with TP53. Br. J. Haematol. 2021, 195, 655–656. [Google Scholar] [CrossRef]
- Fry, E.A.; Inoue, K. C-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65. [Google Scholar] [CrossRef]
- Dúckaa, M.; Kučeríková, M.; Trčka, F.; Červinka, J.; Biglieri, E.; Šmarda, J.; Borsig, L.; Beneš, P.; Knopfová, L. C-Myb Interferes with Inflammatory IL1α-NF-ΚB Pathway in Breast Cancer Cells. Neoplasia 2021, 23, 326–336. [Google Scholar] [CrossRef]
- Inder, S.; O’Rourke, S.; McDermott, N.; Manecksha, R.; Finn, S.; Lynch, T.; Marignol, L. The Notch-3 Receptor: A Molecular Switch to Tumorigenesis? Cancer Treat. Rev. 2017, 60, 69–76. [Google Scholar] [CrossRef]
- Zhu, C.; Ho, Y.J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.S.; Lowe, S.W.; Pajvani, U.B. Notch Activity Characterizes a Common Hepatocellular Carcinoma Subtype with Unique Molecular and Clinicopathologic Features. J. Hepatol. 2021, 74, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt Signaling Pathway in Development and Cancer. J. Physiol. Pharmacol. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Colozza, G.; Koo, B. Ub and Dub of RNF43/ZNRF3 in the WNT Signalling Pathway. EMBO Rep. 2021, 22, e52970. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yasunaga, J.; Fan, J.; Yanagawa, S.; Matsuoka, M. HTLV-1 BZIP Factor Dysregulates the Wnt Pathways to Support Proliferation and Migration of Adult T-Cell Leukemia Cells. Oncogene 2013, 32, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Chihara, Y.; Kobayashi, S.; Iwanaga, M.; Utsunomiya, A.; Watanabe, T.; Uchimaru, K. Overexpression of Aberrant Wnt5a and Its Effect on Acquisition of Malignant Phenotypes in Adult T-Cell Leukemia/Lymphoma (ATL) Cells. Sci. Rep. 2021, 11, 4114. [Google Scholar] [CrossRef]
- Colli, L.M.; Saggioro, F.; Serafini, L.N.; Camargo, R.C.; Machado, H.R.; Moreira, A.C.; Antonini, S.R.; de Castro, M. Components of the Canonical and Non-Canonical Wnt Pathways Are Not Mis-Expressed in Pituitary Tumors. PLoS ONE 2013, 8, e62424. [Google Scholar] [CrossRef]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct Structural Classes of Activating FOXA1 Alterations in Advanced Prostate Cancer. Nature 2019, 571, 413. [Google Scholar] [CrossRef]
- Basham, K.J.; Rodriguez, S.; Turcu, A.F.; Lerario, A.M.; Logan, C.Y.; Rysztak, M.R.; Gomez-Sanchez, C.E.; Breault, D.T.; Koo, B.K.; Clevers, H.; et al. A ZNRF3-Dependent Wnt/β-Catenin Signaling Gradient Is Required for Adrenal Homeostasis. Genes Dev. 2019, 33, 209–220. [Google Scholar] [CrossRef]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and Noncanonical Wnts Use a Common Mechanism to Activate Completely Unrelated Coreceptors. Genes Dev. 2010, 24, 2517. [Google Scholar] [CrossRef]
- Flores-Hernández, E.; Velázquez, D.M.; Castañeda-Patlán, M.C.; Fuentes-García, G.; Fonseca-Camarillo, G.; Yamamoto-Furusho, J.K.; Romero-Avila, M.T.; García-Sáinz, J.A.; Robles-Flores, M. Canonical and Non-Canonical Wnt Signaling Are Simultaneously Activated by Wnts in Colon Cancer Cells. Cell. Signal. 2020, 72, 109636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The Role of TGF-β/SMAD4 Signaling in Cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. TGFβ-Directed Therapeutics: 2020. Pharmacol. Ther. 2021, 217, 107666. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ling, L.; Van Dam, H.; Zhou, F.; Zhang, L. TGF-β Signaling in Cancer Metastasis. Acta Biochim. Biophys. Sin. Shanghai 2018, 50, 121–132. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; De Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 Interactome Reveals That TGFβ Controls m 6 A MRNA Methylation in Pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- MaruYama, T.; Chen, W.J.; Shibata, H. TGF-β and Cancer Immunotherapy. Biol. Pharm. Bull. 2022, 45, 155–161. [Google Scholar] [CrossRef]
- Zhao, X.; Li, D.; Qiu, Q.; Jiao, B.; Zhang, R.; Liu, P.; Ren, R. Zfyve16 Regulates the Proliferation of B-Lymphoid Cells. Front. Med. 2018, 12, 559–565. [Google Scholar] [CrossRef]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘Hallmarks of Cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Y.; Yang, C.; Simeone, D.M.; Sun, T.T.; DeGraff, D.J.; Tang, M.S.; Zhang, Y.; Wu, X.R. Dominant Role of CDKN2B/P15INK4B of 9p21.3 Tumor Suppressor Hub in Inhibition of Cell-Cycle and Glycolysis. Nat. Commun. 2021, 12, 2047. [Google Scholar] [CrossRef] [PubMed]
- Fauquenoy, S.; Robette, G.; Kula, A.; Vanhulle, C.; Bouchat, S.; Delacourt, N.; Rodari, A.; Marban, C.; Schwartz, C.; Burny, A.; et al. Repression of Human T-Lymphotropic Virus Type 1 Long Terminal Repeat Sense Transcription by Sp1 Recruitment to Novel Sp1 Binding Sites. Sci. Rep. 2017, 7, srep43221. [Google Scholar] [CrossRef] [PubMed]
- Jabareen, A.; Suleman, M.; Abu-Jaafar, A.; Huleihel, M. Different Molecular Mechanisms of HTLV-1 and HIV LTR Activation by TPA. Biochem. Biophys. Res. Commun. 2018, 500, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wu, R.H.; Zhou, H.L.; Li, Z.M.; Kou, D.; Deng, Z.; Dong, M.; Chen, L.H. TGIF2 Promotes Cervical Cancer Metastasis by Negatively Regulating FCMR. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Wu, D.; Fujiwara, K.; Ishizuka, Y.; Oguni, A.; Tokunaga, T.; Takayama, T.; Soma, M.; Fukuda, N.; Ozaki, T.; et al. Knockdown of E2F5 Induces Cell Death via the TP53-dependent Pathway in Breast Cancer Cells Carrying Wild-type TP53. Oncol. Rep. 2020, 44, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.S.; Wotton, D.; Massagué, J. Epidermal Growth Factor Signaling via Ras Controls the Smad Transcriptional Co-Repressor TGIF. EMBO J. 2001, 20, 128–136. [Google Scholar] [CrossRef]
- Jin, Z.; Zhou, S.; Ye, H.; Jiang, S.; Yu, K.; Ma, Y. The Mechanism of SP1/P300 Complex Promotes Proliferation of Multiple Myeloma Cells through Regulating IQGAP1 Transcription. Biomed. Pharmacother. 2019, 119, 109434. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Gao, H.; Xu, B.; Zhai, W.; Li, J.; Zhang, C. Elevated Expression of TGIF Is Involved in Lung Carcinogenesis. Tumour Biol. 2015, 36, 9223–9231. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, L.; Li, J.; Li, L.; Wang, H.; Yang, H. Long-Term Cadmium Exposure Promoted Breast Cancer Cell Migration and Invasion by up-Regulating TGIF. Ecotoxicol. Environ. Saf. 2019, 175, 110–117. [Google Scholar] [CrossRef]
- Chen, C.R.; Kang, Y.; Siegel, P.M.; Massagué, J. E2F4/5 and P107 as Smad Cofactors Linking the TGFbeta Receptor to c-Myc Repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef]
- Thurlings, I.; De Bruin, A. E2F Transcription Factors Control the Roller Coaster Ride of Cell Cycle Gene Expression. Methods Mol. Biol. 2016, 1342, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The Broken Cycle: E2F Dysfunction in Cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Satou, Y.; Sugata, K.; Miyazato, P.; Green, P.L.; Imamura, T.; Matsuoka, M. HTLV-1 BZIP Factor Enhances TGF-β Signaling through P300 Coactivator. Blood 2011, 118, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Guérit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.B. PDGF Receptor Mutations in Human Diseases. Cell. Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef]
- Liu, W.N.; Yan, M.; Chan, A.M. A Thirty-Year Quest for a Role of R-Ras in Cancer: From an Oncogene to a Multitasking GTPase. Cancer Lett. 2017, 403, 59–65. [Google Scholar] [CrossRef]
- Xiao, R.; Shi, L.; Yang, T.; Zhang, M.; Wang, H.; Mai, S. Identification of RRAS Gene Related to Nasopharyngeal Carcinoma Based on Pathway and Network-Based Analyses. Transl. Cancer Res. 2019, 8, 664–675. [Google Scholar] [CrossRef]
- Weber, S.M.; Carroll, S.L. The Role of R-Ras Proteins in Normal and Pathologic Migration and Morphologic Change. Am. J. Pathol. 2021, 191, 1499–1510. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm. Sin. B 2019, 9, 871. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, F.; Xu, D.; Hou, K.; Fang, W.; Li, Y. The Function of RAS Mutation in Cancer and Advances in Its Drug Research. Curr. Pharm. Des. 2019, 25, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Stoppa, G.; Rumiato, E.; Saggioro, D. Ras Signaling Contributes to Survival of Human T-Cell Leukemia/Lymphoma Virus Type 1 (HTLV-1) Tax-Positive T-Cells. Apoptosis 2012, 17, 219. [Google Scholar] [CrossRef] [PubMed]
- Vajente, N.; Trevisan, R.; Saggioro, D. HTLV-1 Tax Protein Cooperates with Ras in Protecting Cells from Apoptosis. Apoptosis 2009, 14, 153–163. [Google Scholar] [CrossRef]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the Era of Immune Checkpoint Inhibitors: Friend or Foe? Nat. Rev. Rheumatol. 2021, 17, 213. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef]
- Li, J.; Liu, N.; Tang, L.; Yan, B.; Chen, X.; Zhang, J.; Peng, C. The Relationship between TRAF6 and Tumors. Cancer Cell Int. 2020, 20, 429. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Rauch, D.A.; Ratner, L. Targeting HTLV-1 Activation of NFκB in Mouse Models and ATLL Patients. Viruses 2011, 3, 886. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Giam, C.Z. NF-ΚB Signaling Mechanisms in HTLV-1-Induced Adult T-Cell Leukemia/Lymphoma. FEBS J. 2018, 285, 3324. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Type of Samples Analyzed | Proposed Cellular Pathways | Expression Levels | Reference |
|---|---|---|---|---|
| miR-199a-3p | Samples of ATLL patients and asymptomatic HTLV-1 carriers | NR | Downregulated when comparing ATL patients to asymptomatic HTLV-1 carriers, but upregulated when comparing ATL patients to healthy controls | [46] |
| miR-26a-5p | Predicted to target ABHD2, HMGA1, EP400, CDK8, ZNF608, KPNA6, and ZSWIM6 | |||
| miR-199b-3p | NR | |||
| miR-150-5p | Predicted to target ADIPOR2, SP1, ZEB1, EGR2, and CBL | |||
| let-7d-3p | NR | |||
| miR-155-5p | Predicted to target MORC3, TRIM32, SMAD2, and TP53INP1 | |||
| miR-26b-5p | Predicted to target CREBZF, USP3, KPNA6, and RAP2C | |||
| miR-222-3p | Predicted to target PANK3, TLE3, ZFYVE16, PHACTR4, and SUN2 | |||
| miR-181b-5p | Predicted to target ZNF780B, HEPHL1, ZNF268, ZBTB4, PTBP3, NR6A1, PBX3, CAPRIN2, PHC3, C2orf69, INO80D, CPOX, KPNA1, TNPO1, PTEN, GSKIP, ARF6, and MPP5 | |||
| miR-30e-3p | NR | |||
| miR-127 | Samples obtained from HTLV-1 infected patients at the time of diagnosis | NR | Downregulated in HTLV-1 infected patients | [47] |
| miR-136 | ||||
| miR-142-3p | ||||
| miR-221 | ||||
| miR-423-5p | ||||
| let-7b | Upregulated in HTLV-1 infected patients | |||
| miR-29c | ||||
| miR-30c | ||||
| miR-193a-5p | ||||
| miR-885-5p | ||||
| miR-34a | C91PL, MT-2, HUT-102, C8166, ATL-2, and ED40515(−); Samples from ATLL patients | A transcriptional target of p53, NF-kB, Tap73, and ELK; Targets many cell proliferation and survival pathways such as MYC, MYCN, MET, CCDN1, CDK6, BCL2, and NOTCH1; May modulate expression of tumor suppressor genes | Upregulated both in cell lines, except ED40515(−), and in patient samples when compared to PBMC of healthy donors | [48] |
| miR-150 | MT-4, MT-2, C8166, C91PL, Jurkat, MT-1, ATL-T, ED-40515(−), ALT-25, ATL-43T, LMY1, and ATL-55T; Samples from ATLL patients | Inhibition of STAT1 expression and suppression of STAT1-dependent genes | Downregulated in HTLV-1 infected and ATL-like cell lines | [49] |
| miR-223 | ||||
| miR-17 | CD4+ and CD8+ T cells from HTLV-1 infected individuals and healthy donors | Upregulated in an HBZ-dependent manner; Trigger cell proliferation and genomic instability through inhibition of OBFC2A-hSSB2 pathway | Upregulated in CD4+ infected clones when compared to uninfected CD4+ clones | [50] |
| miR-21 | ||||
| miR-23b | Upregulated in an HBZ-dependent manner | |||
| miR-27b | ||||
| miR-34a-5p | C91PL and MT-2 | Regulator of cell proliferation and survival in a p53-dependent manner; Its upregulation in other virus-associated malignancies suggests diverse cellular effects depending on context | Upregulated in HTLV-1 infected cell lines | [51] |
| miR-150-5p | Target oncogenes c-Myb and NOTCH-3; Antiproliferative and proapoptotic effects on B-lymphoma, T-ALL, and NK cell lines | Downregulated in HTLV-1 infected cell lines | ||
| miR-146b-5p | Potential activity over TRAF6, IRAK1, FADD, and CXCR4 | |||
| miR-155 | MT-2, MT-4, C5/MJ, SLB-1, HUT-102, MT-1, and ED-40515(−), Jurkat, MOLT-4, CCRF-CEM, and JPX-9 | Upregulation of miR-155 by Tax through activation of NF-kB and AP-1; Potential inhibition of transcriptional repressors BACH1 and HIVEP2 | Upregulated in HTLV-1 infected cell lines | [52] |
| miR-149 | Jurkat and MT-2 | Act upon histone acetyltransferases p300 and p/CAF, regulating chromatin remodeling | Downregulated in HTLV-1 infected cell lines when compared to Jurkat | [53] |
| miR-873 | ||||
| miR-31 | Samples of ATLL patients | Regulated by Polycomb proteins activity; Inhibits NF-kB-inducing kinase (NIK) | Downregulated in ATL samples when compared to healthy donors | [54] |
| miR-146a | MT-2, MT-4, C5/MJ, SLB-1, MT-1, ED-40515(−), HUT-102, Jurkat, MOLT-4, CCRF-CEM, and JPX-9 | Upregulation of miR-146a by Tax through activation of NF-kB; Enhances cell growth through undetermined mechanisms; Able to target TRAF6 and IRAK1 | Upregulated in HTLV-1 infected cells when compared to non-infected cell lines | [55] |
| miR-181a | C8166, MT-2, MT-4, HUT102, LAF, MUO4; Samples from ATLL patients | Favors B cell differentiation and regulates T cell receptor signaling | Downregulated in HTLV-1 infected cell lines and ATL patient samples | [56] |
| miR-132 | Involved in innate immunity | |||
| miR-125a | Involved in innate immunity and regulation of regulatory T cells functions | |||
| miR-155 | Upregulated through NF-kB and JNK pathways; Regulates dendritic and T cell interactions as well as T helper cells differentiation | Upregulated in HTLV-1 infected cell lines and ATL patient samples | ||
| miR-142-3p | Induces differentiation towards T cell lymphopoiesis | |||
| miR-150 | Regulates differentiation of B and T cell lineages | Downregulated in HTLV-1 infected cell lines, but upregulated in ATL patient samples | ||
| miR-223 | Induces differentiation towards T cell lymphopoiesis | |||
| miR-142-5p | Induces differentiation towards T cell lymphopoiesis | Upregulated ATL patient samples | ||
| miR-146b | Involved in innate immunity | Downregulated ATL patient samples | ||
| miR-223 | Jurkat, HuT-78, CEM, HuT-102, StEd, ATL-3, PaBe, JuanaW, Champ, C91-PL, MT-2, Abgho, Nilu, Eva, Xpos, and Tesi | NR | Downregulated in HTLV-1 infected and ATL-derived cell lines | [57] |
| miR-21 | Predicted binding sites to a cohort of regulatory genes through in silico analysis | Upregulated in HTLV-1 infected and ATL-derived cell lines | ||
| miR-24 | ||||
| miR-155 | ||||
| miR-146a | Upregulation of miR-146a by Tax through activation of NF-kB; Predicted binding sites to a cohort of regulatory genes through in silico analysis | |||
| miR-93 | MT-1. ATL55T, ATL-2, ATL48T, TLOM1, ED, 43T, MT-4; Samples from ATLL patients | Inhibition of tumor suppressor TP53INP1 | Upregulated in HTLV-1 infected cell lines and ATL patient samples | [58] |
| miR-130b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, C.B.; da Cunha, L.S.; Maués, J.H.d.S.; Pessoa, F.M.C.d.P.; de Oliveira, M.B.; Ribeiro, R.M.; Lopes, G.S.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Khayat, A.S.; et al. Role of miRNAs in Human T Cell Leukemia Virus Type 1 Induced T Cell Leukemia: A Literature Review and Bioinformatics Approach. Int. J. Mol. Sci. 2022, 23, 5486. https://doi.org/10.3390/ijms23105486
Machado CB, da Cunha LS, Maués JHdS, Pessoa FMCdP, de Oliveira MB, Ribeiro RM, Lopes GS, de Moraes Filho MO, de Moraes MEA, Khayat AS, et al. Role of miRNAs in Human T Cell Leukemia Virus Type 1 Induced T Cell Leukemia: A Literature Review and Bioinformatics Approach. International Journal of Molecular Sciences. 2022; 23(10):5486. https://doi.org/10.3390/ijms23105486
Chicago/Turabian StyleMachado, Caio Bezerra, Leidivan Sousa da Cunha, Jersey Heitor da Silva Maués, Flávia Melo Cunha de Pinho Pessoa, Marcelo Braga de Oliveira, Rodrigo Monteiro Ribeiro, Germison Silva Lopes, Manoel Odorico de Moraes Filho, Maria Elisabete Amaral de Moraes, André Salim Khayat, and et al. 2022. "Role of miRNAs in Human T Cell Leukemia Virus Type 1 Induced T Cell Leukemia: A Literature Review and Bioinformatics Approach" International Journal of Molecular Sciences 23, no. 10: 5486. https://doi.org/10.3390/ijms23105486
APA StyleMachado, C. B., da Cunha, L. S., Maués, J. H. d. S., Pessoa, F. M. C. d. P., de Oliveira, M. B., Ribeiro, R. M., Lopes, G. S., de Moraes Filho, M. O., de Moraes, M. E. A., Khayat, A. S., & Moreira-Nunes, C. A. (2022). Role of miRNAs in Human T Cell Leukemia Virus Type 1 Induced T Cell Leukemia: A Literature Review and Bioinformatics Approach. International Journal of Molecular Sciences, 23(10), 5486. https://doi.org/10.3390/ijms23105486