
A Comprehensive Assessment of Genetic and Epigenetic Alterations Identifies Frequent Variations Impacting Six Prototypic SCF Complex Members


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
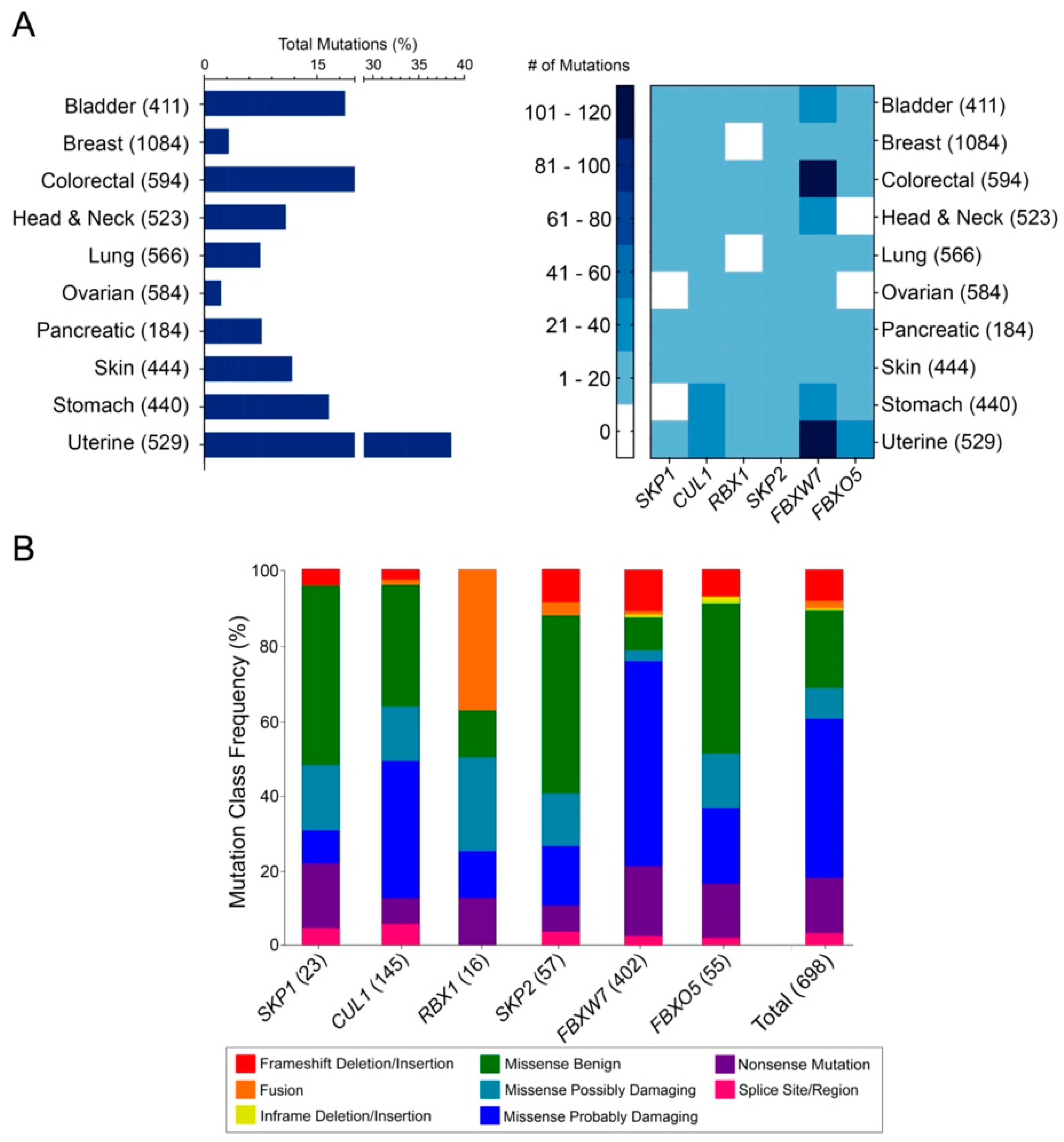
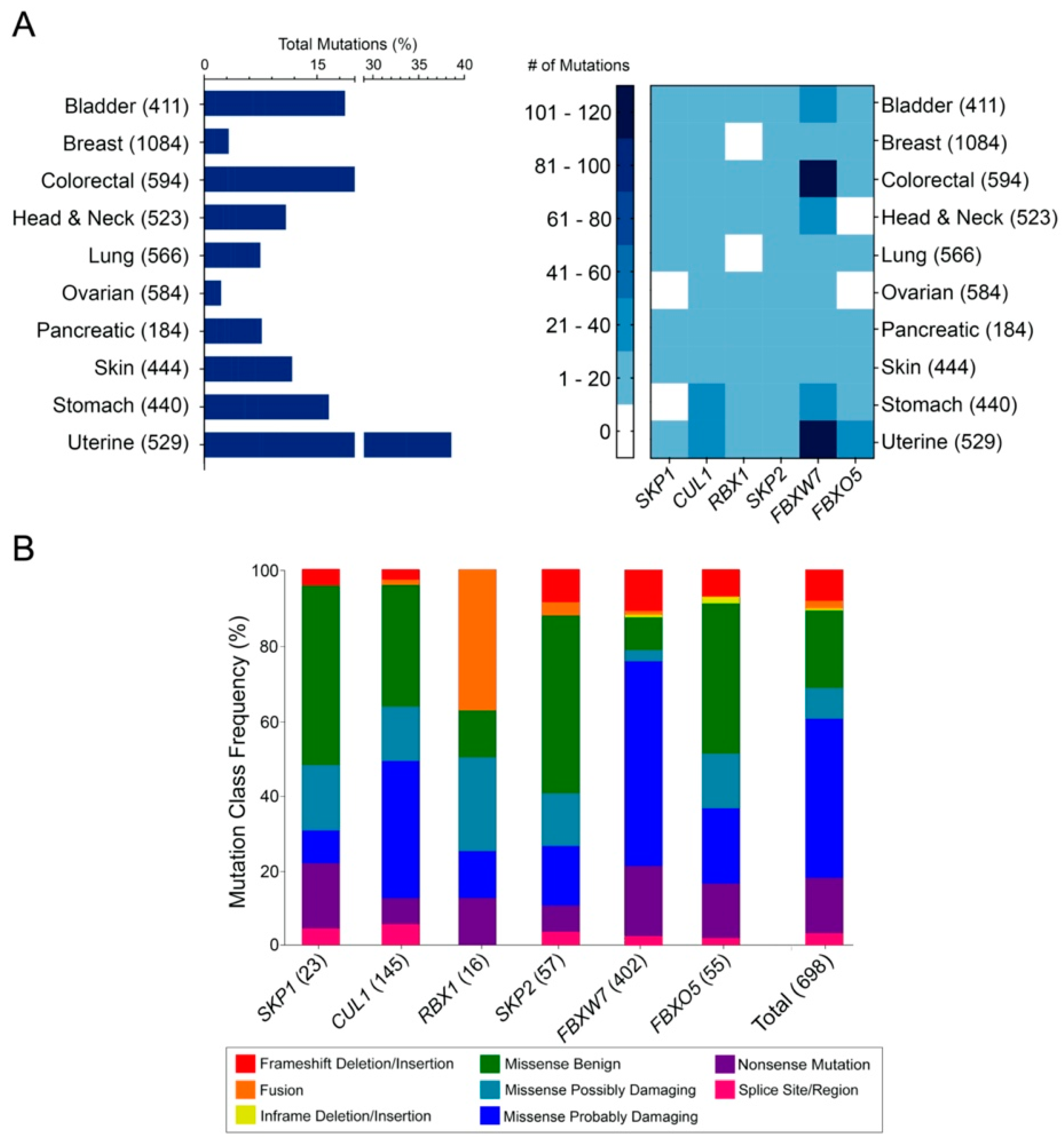
2.1. Genes Encoding SCF Complex Members Are Mutated Frequently in Cancer
2.2. The Distributions of Encoded SCF Complex Alterations Are Consistent with a Tumor Suppressor Mutational Load
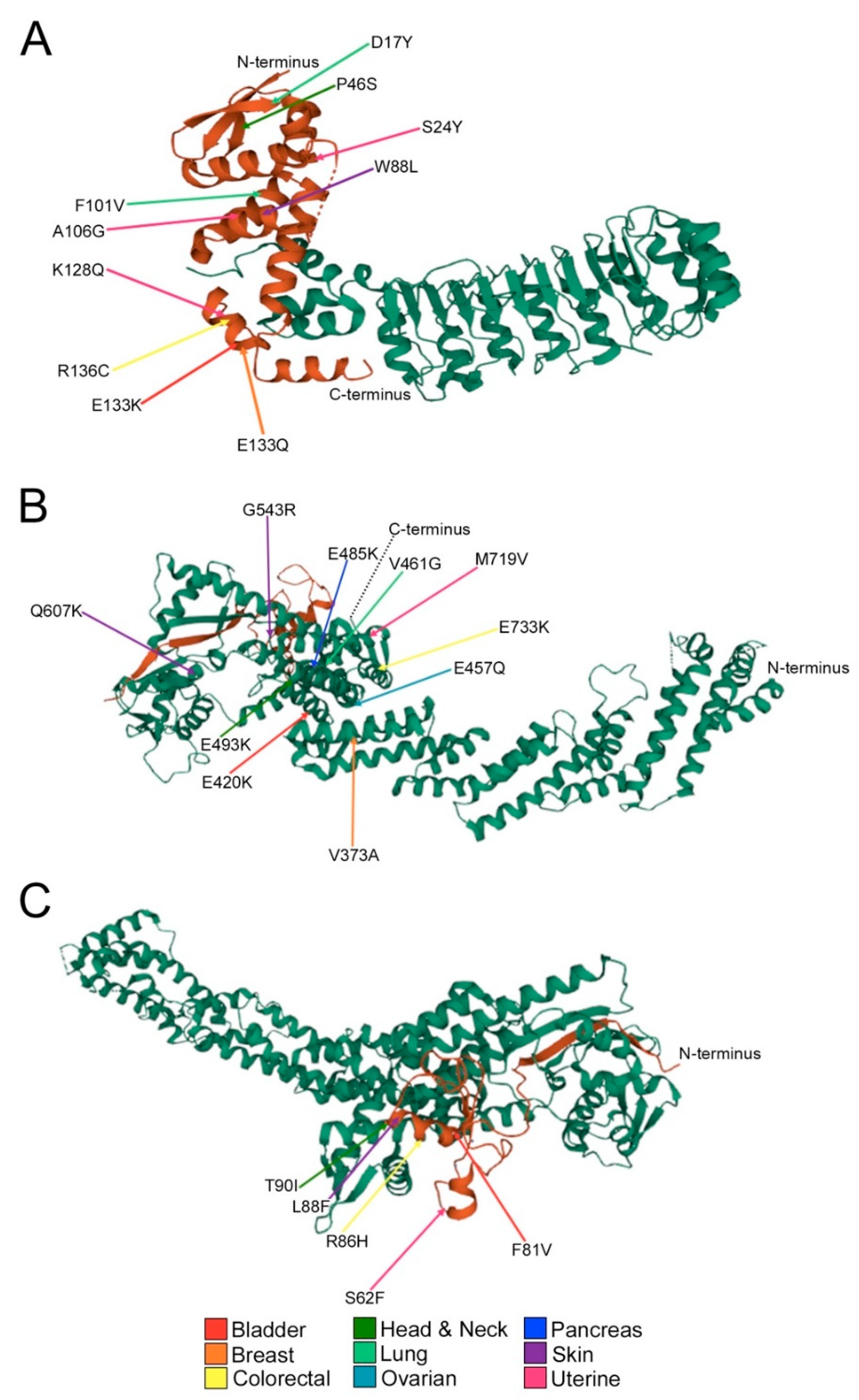
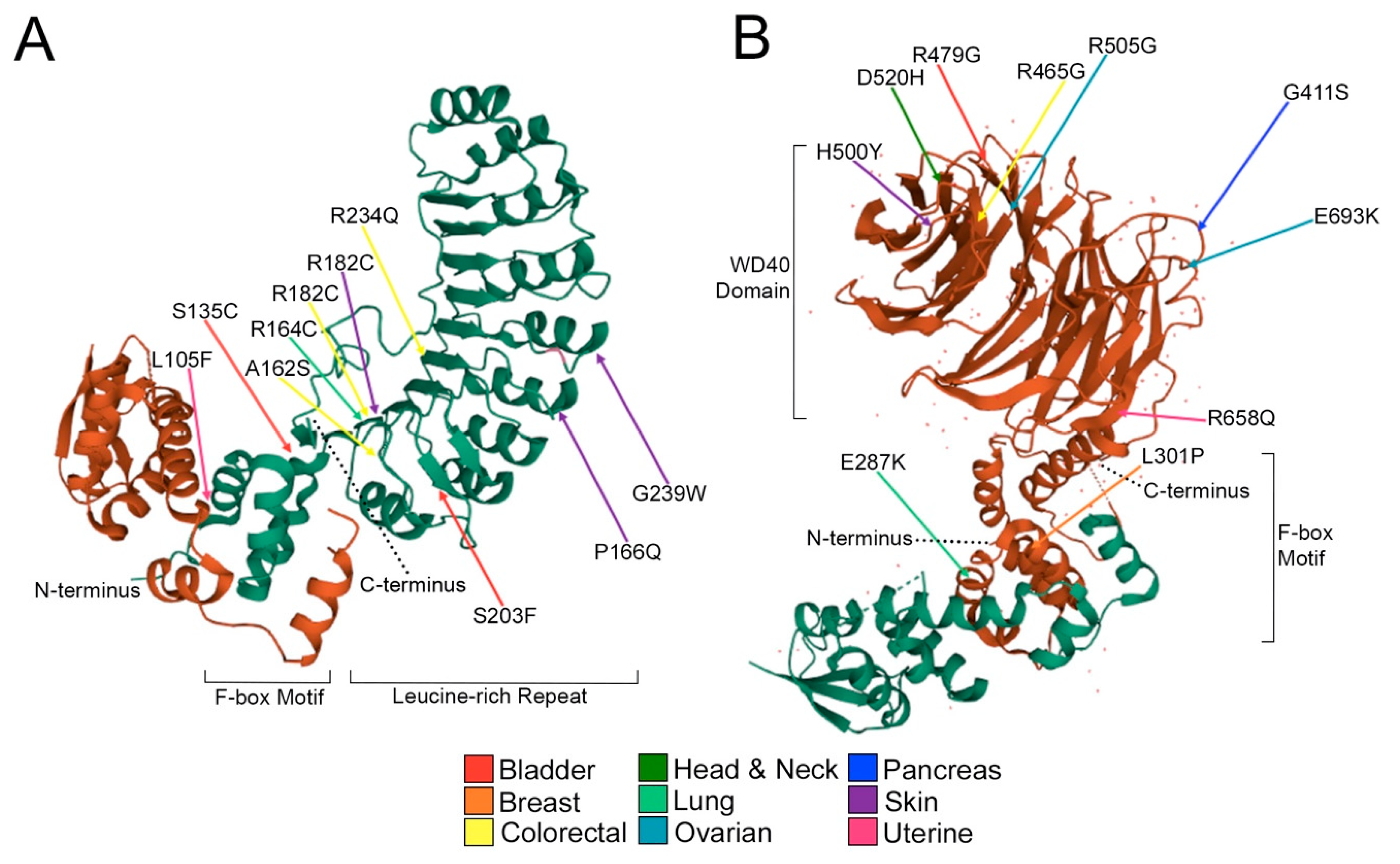
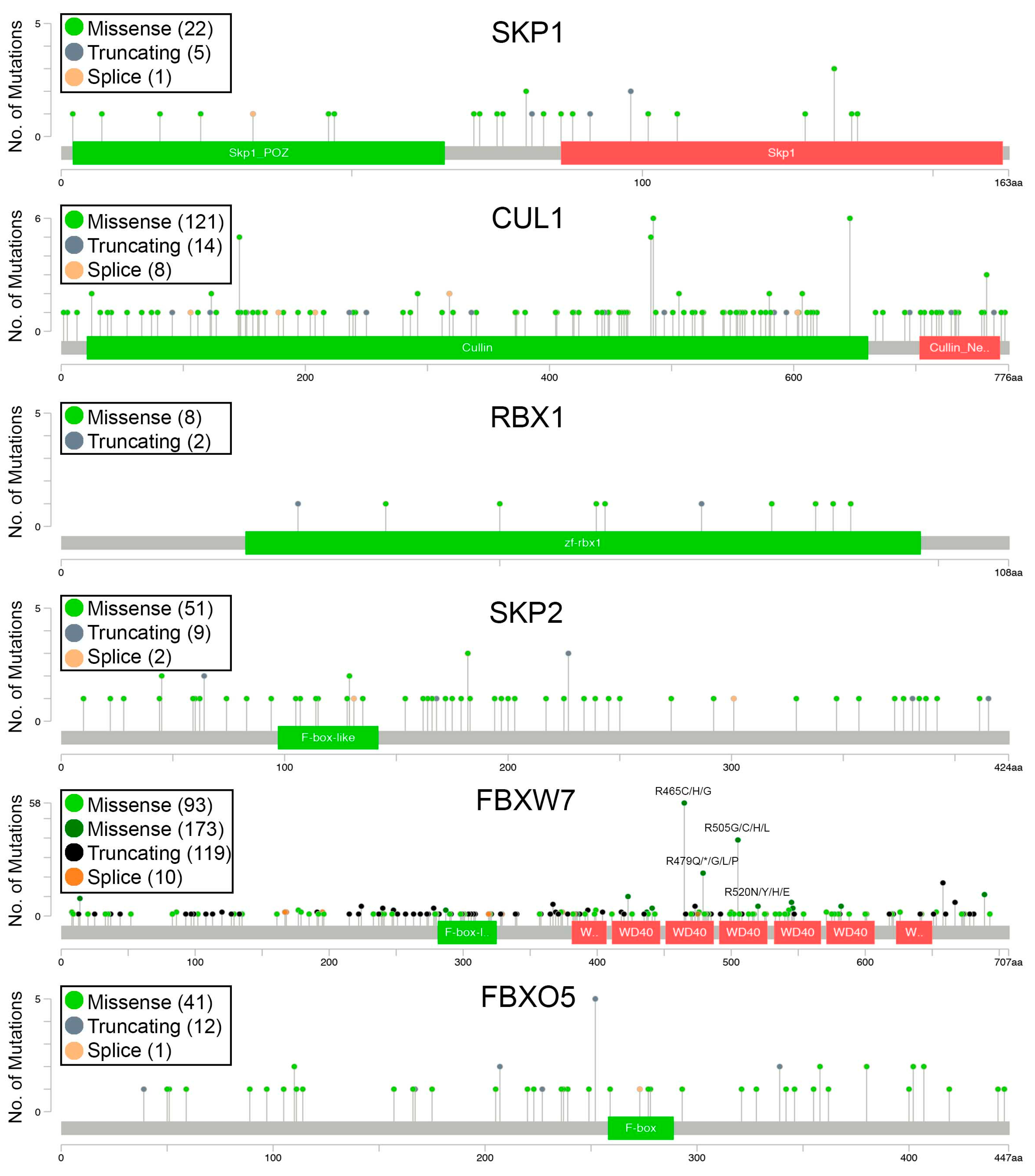
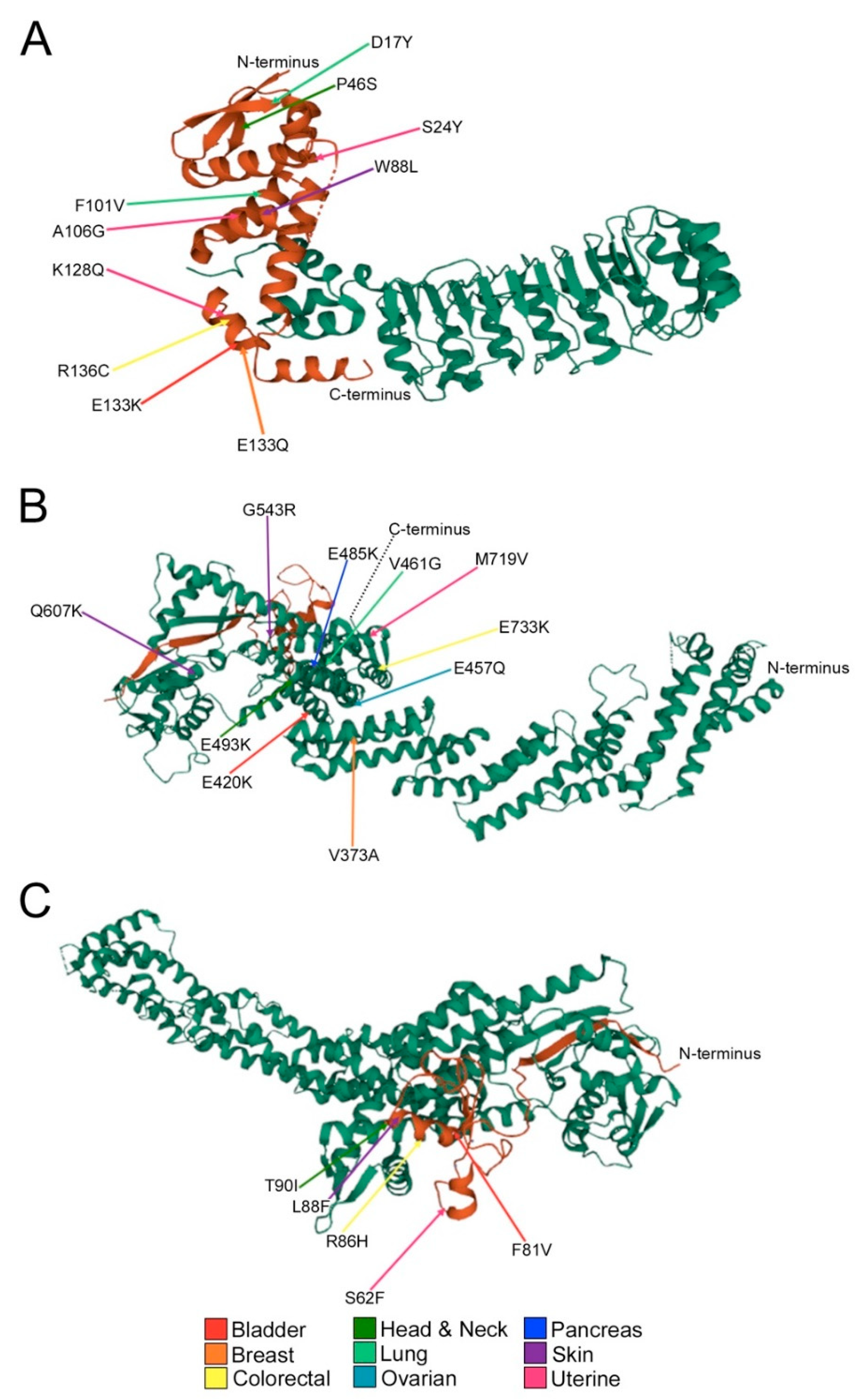
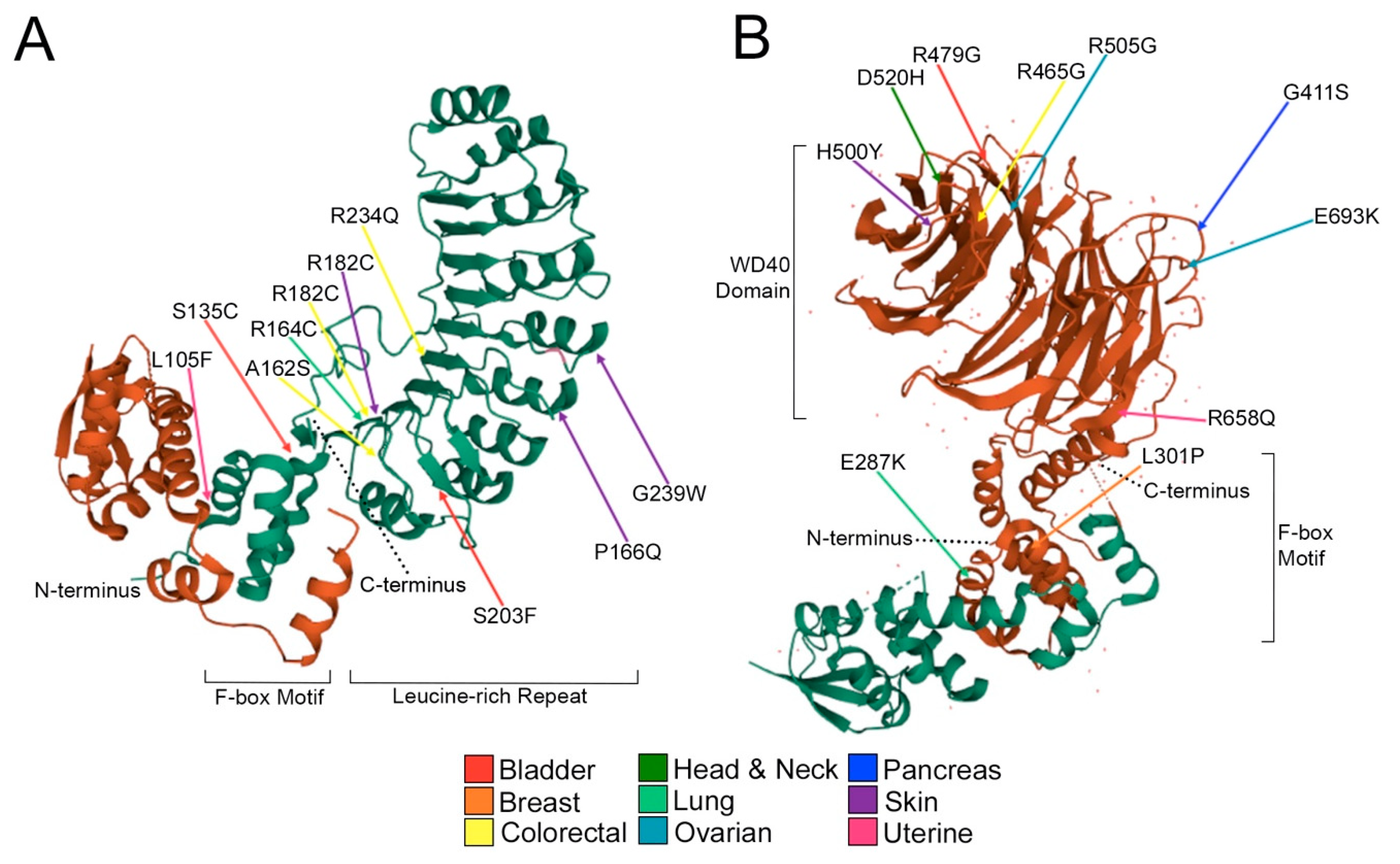
2.3. Encoded Alterations in SCF Complex Members May Adversely Impact Protein Structure
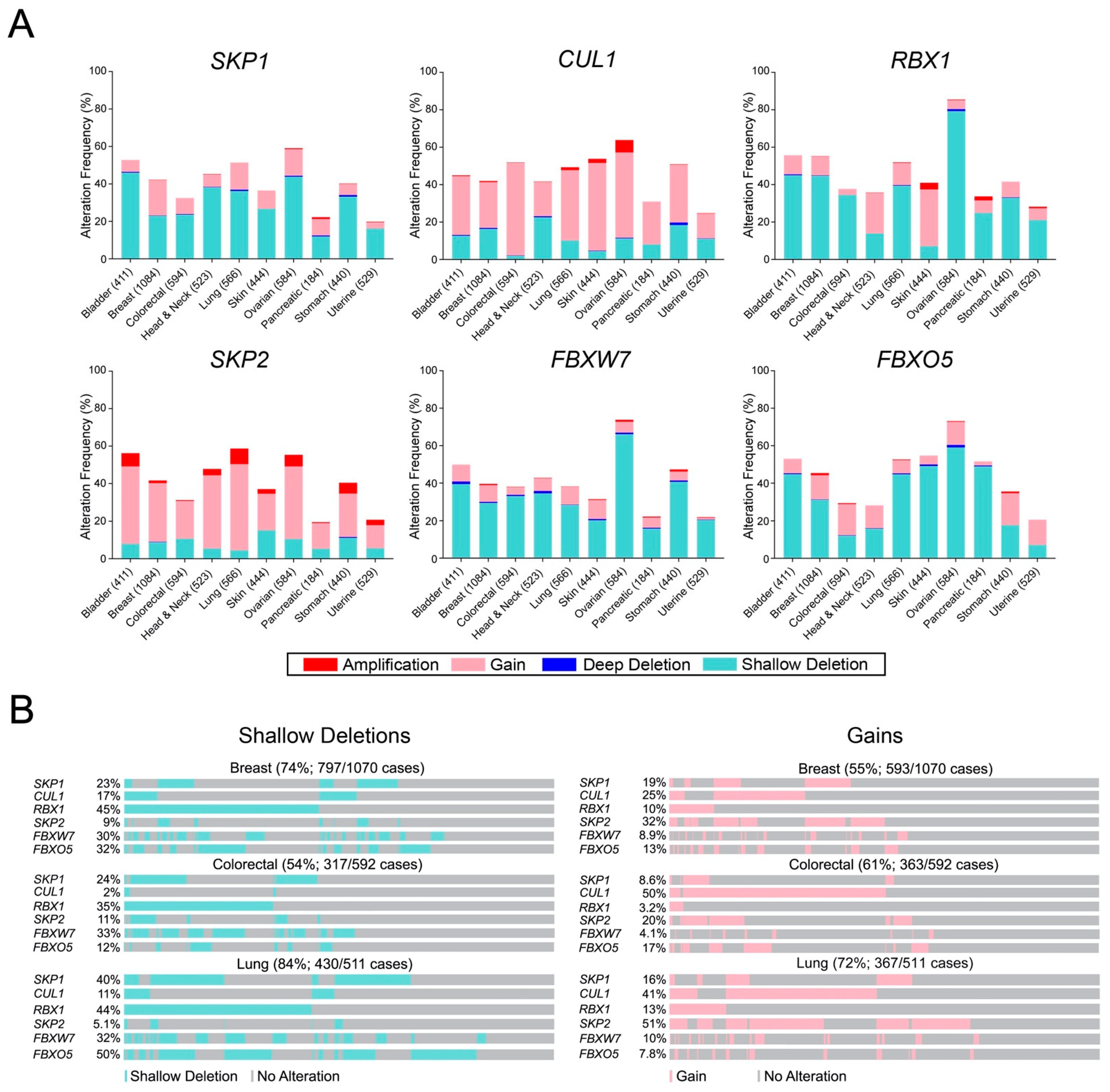
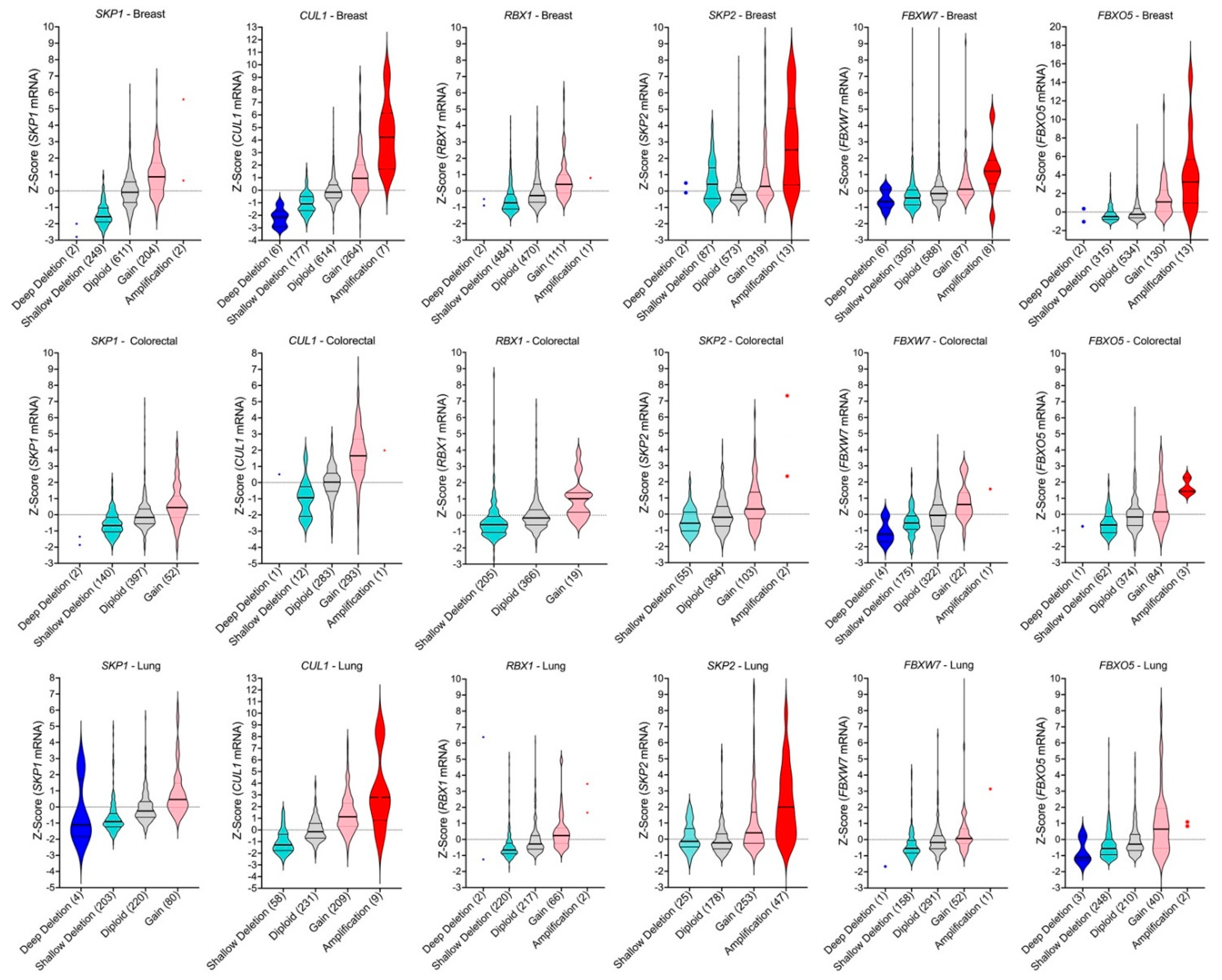
2.4. SCF Complex Members Exhibit Frequent Copy Number Alterations in Cancer
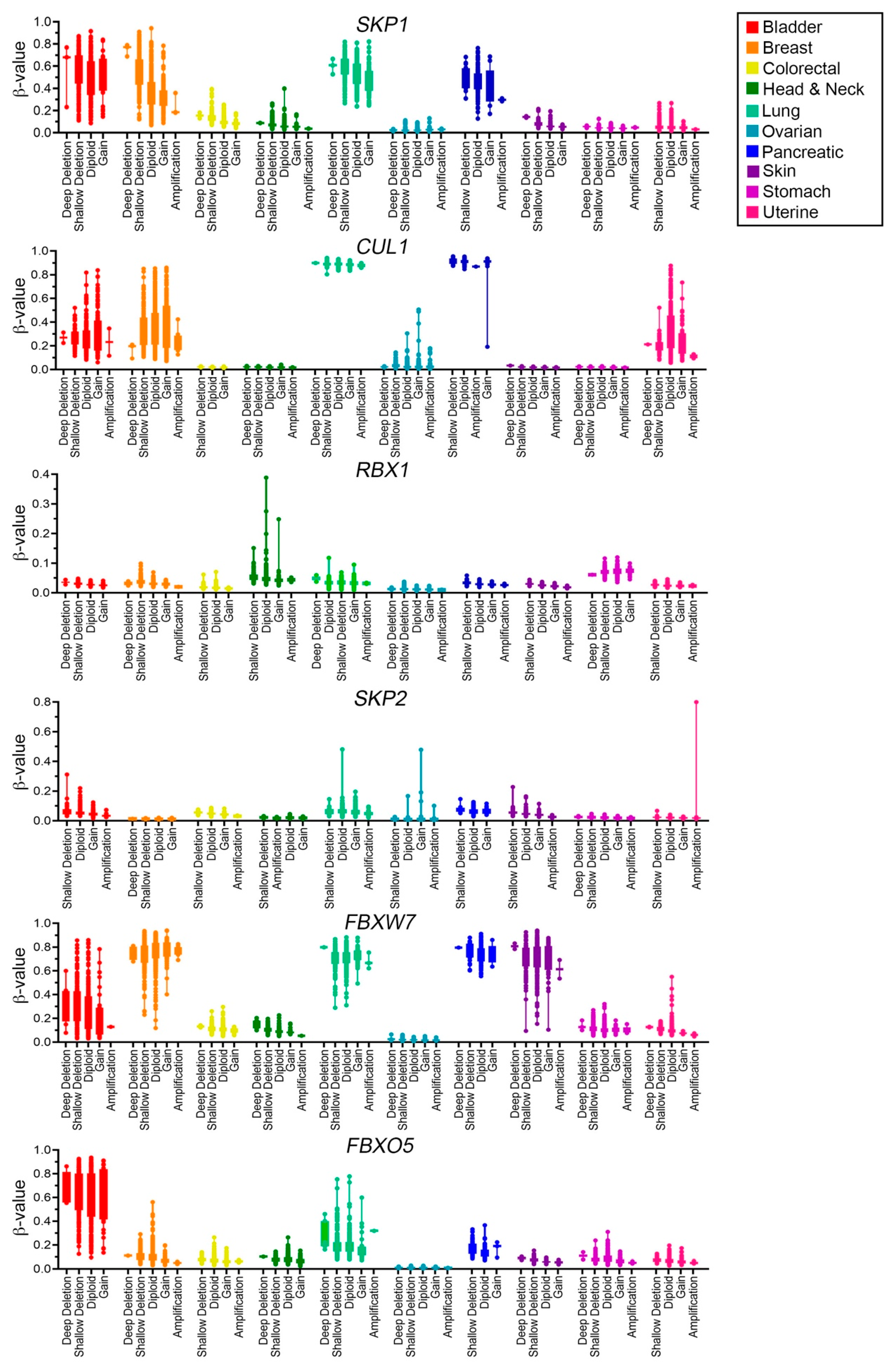
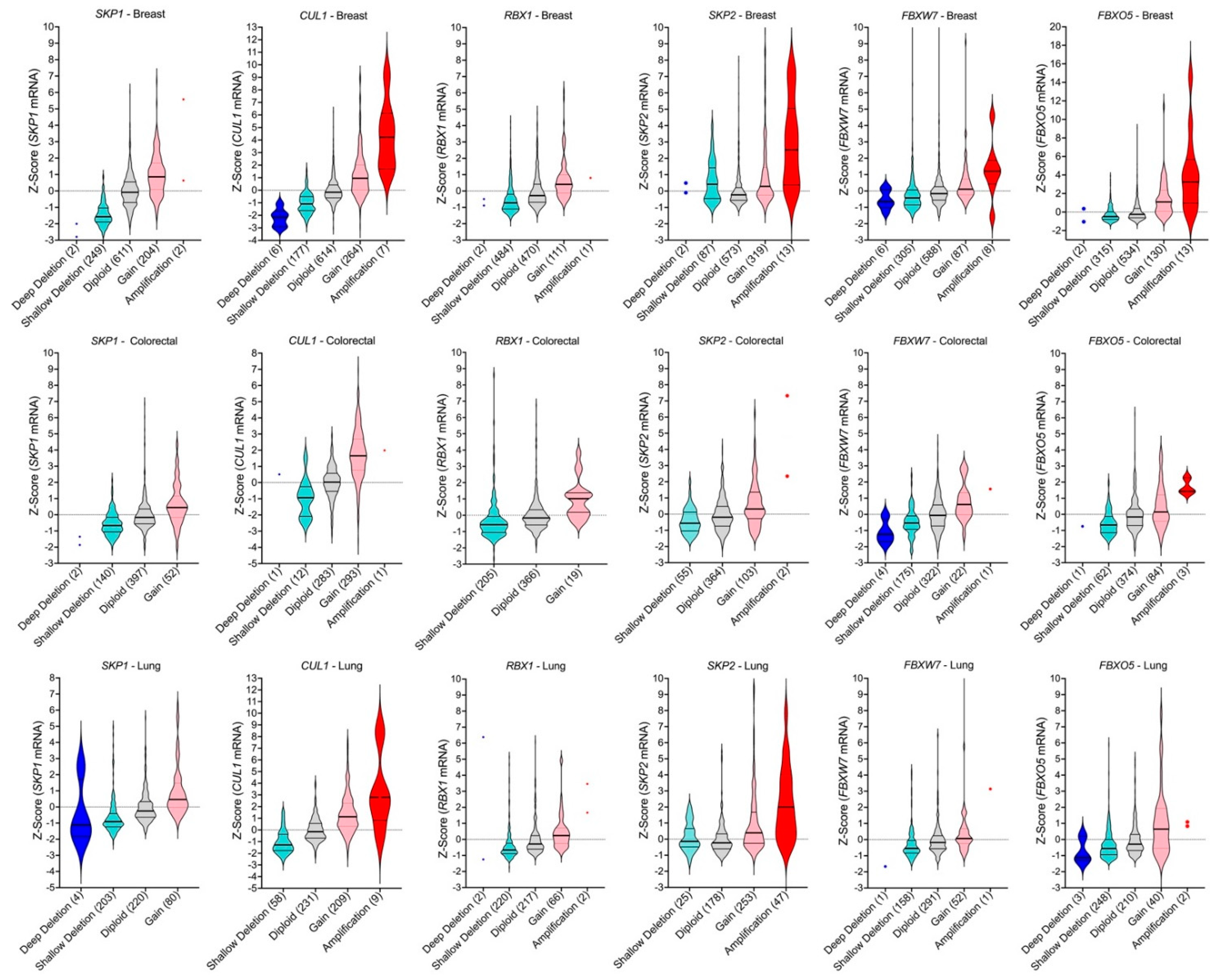
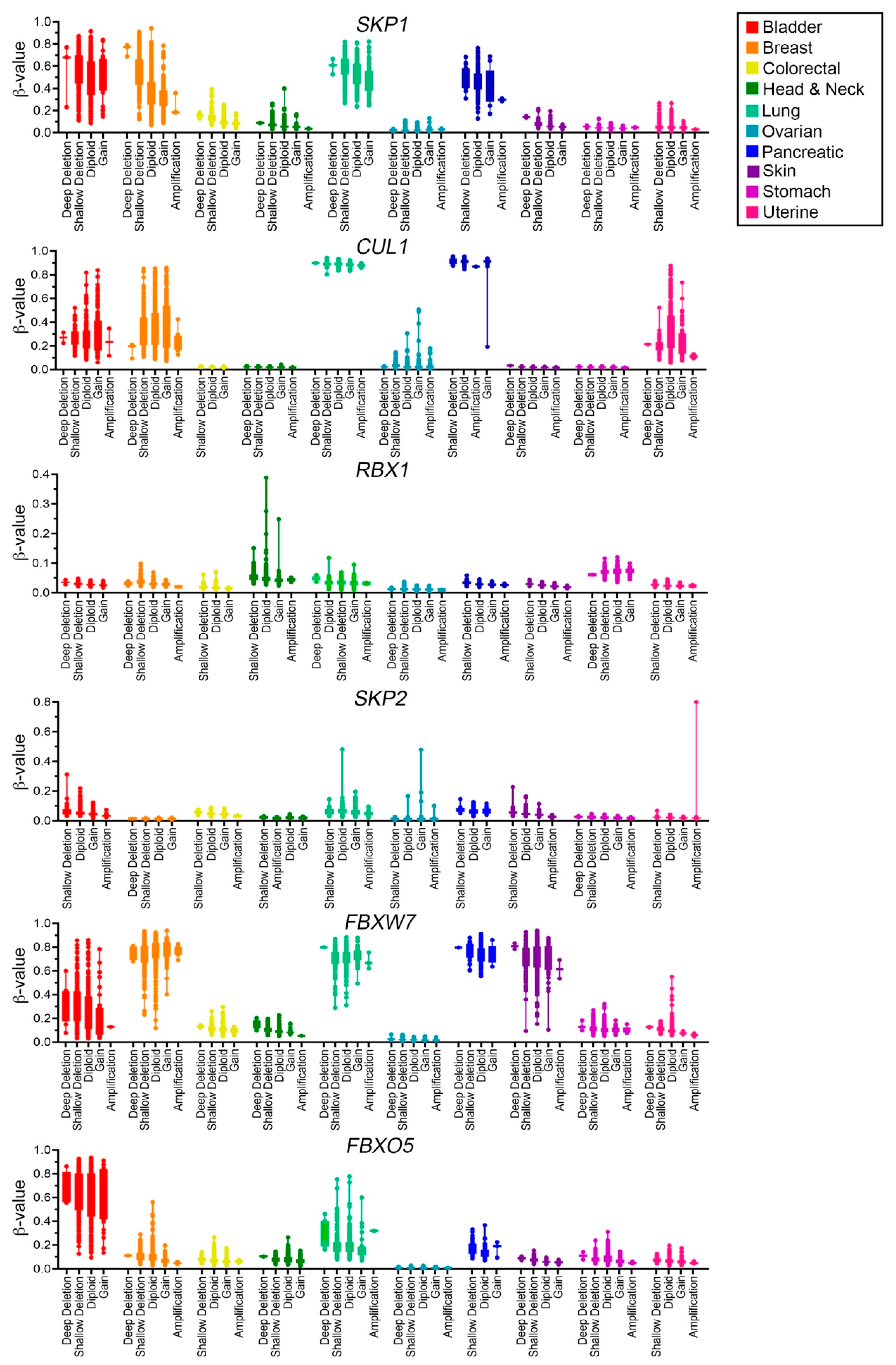
2.5. SCF Complex Members Are Differentially Methylated in Cancer
3. Discussion
4. Materials and Methods
4.1. Genomic Datasets and Data Collection
4.2. Assessing the Frequency, Distribution and Predicted Functional Impact of Encoded SCF Complex Mutations
4.3. Assessing the Predicted Structural Impact of Encoded SCF Complex Alterations
4.4. Gene Copy Number Alteration and mRNA Expression Analyses
4.5. Assessing the Methylation Status of SCF Complex Members
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069–2081. [Google Scholar] [CrossRef] [Green Version]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.M.; Olszewski, J.L.; Tron, A.E.; Hammel, M.; Lambert, L.J.; Waddell, M.B.; Mittag, T.; DeCaprio, J.A.; Schulman, B.A. Structure of a Glomulin-RBX1-CUL1 Complex: Inhibition of a RING E3 Ligase through Masking of Its E2-Binding Surface. Mol. Cell 2012, 47, 371–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshaies, R.J. SCF and Cullin/RING H2-Based Ubiquitin Ligases. Annu. Rev. Cell Dev. Biol. 1999, 15, 435–467. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, D.; Ng, H.F.; Sharifi, H.R.; Mahmoudi, S.; Cerrato, V.S.; Fredlund, E.; Magnusson, K.; Nilsson, H.; Malyukova, A.; Rantala, J.; et al. CDK-mediated activation of the SCFFBXO28 ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol. Med. 2013, 5, 999–1018. [Google Scholar] [CrossRef]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 regulates Myc protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Zhang, Q.; Spears, E.; Boone, D.N.; Li, Z.; Gregory, M.A.; Hanna, S.R. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc. Natl. Acad. Sci. USA 2013, 110, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Bungsy, M.; Palmer, M.C.L.; Jeusset, L.M.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced RBX1 expression induces chromosome instability and promotes cellular transformation in high-grade serous ovarian cancer precursor cells. Cancer Lett. 2021, 500, 194–207. [Google Scholar] [CrossRef]
- Lepage, C.C.; Palmer, M.C.L.; Farrell, A.C.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced SKP1 and CUL1 expression underlies increases in Cyclin E1 and chromosome instability in cellular precursors of high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 1699–1710. [Google Scholar] [CrossRef]
- Palmer, M.C.L.; Neudorf, N.M.; Farrell, A.C.; Razi, T.; Lichtensztein, Z.; McManus, K.J. The F-box protein, FBXO7 is required to maintain chromosome stability in humans. Hum. Mol. Genet. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Baergen, A.K.; Lichtensztejn, Z.; McManus, K.J. Reduced SKP1 expression induces chromosome instability through aberrant cyclin E1 protein turnover. Cancers 2020, 12, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Loda, M.; Cukor, B.; Tam, S.W.; Lavin, P.; Fiorentino, M.; Draetta, G.F.; Jessup, J.M.; Pagano, M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med. 1997, 3, 231–234. [Google Scholar] [CrossRef]
- Joazeiro, C.A.; Wing, S.S.; Huang, H.; Leverson, J.D.; Hunter, T.; Liu, Y.C. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 1999, 286, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Waterman, H.; Levkowitz, G.; Alroy, I.; Yarden, Y. The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J. Biol. Chem. 1999, 274, 22151–22154. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.I.; Hatakeyama, S.; Nakayama, K. Regulation of the cell cycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Biochem. Biophys. Res. Commun. 2001, 282, 853–860. [Google Scholar] [CrossRef]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system - Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.L.; Rutherford, K.A.; Lepage, C.C.; McManus, K.J. The SCF Complex Is Essential to Maintain Genome and Chromosome Stability. Int. J. Mol. Sci. 2021, 22, 8544. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Gabrielsen, M.; Huang, D.T. Single-Turnover RING/U-Box E3-Mediated Lysine Discharge Assays. Methods Mol. Biol. 2018, 1844, 19–31. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Thrower, J.S. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Tetzlaff, M.T.; Yu, W.; Li, M.; Zhang, P.; Finegold, M.; Mahon, K.; Harper, J.W.; Schwartz, R.J.; Elledge, S.J. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc. Natl. Acad. Sci. USA 2004, 101, 3338–3345. [Google Scholar] [CrossRef] [Green Version]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Lu, Y.; Liu, Y.Q.; Su, K.; Zhang, J.; Liu, J.; Zhou, G.B. Skp1: Implications in cancer and SCF-oriented anti-cancer drug discovery. Pharmacol. Res. 2016, 111, 34–42. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, D.C.; Dolios, G.; Wang, R.; Pan, Z.Q. CUL7: A DOC domain-containing cullin selectively binds Skp1·Fbx29 to form an SCF-like complex. Proc. Natl. Acad. Sci. USA 2002, 99, 16601–16606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Schulman, B.A.; Carrano, A.C.; Jeffrey, P.D.; Bowen, Z.; Kinnucan, E.R.; Finnin, M.S.; Elledge, S.J.; Harper, J.W.; Pagano, M.; Pavletich, N.P. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature 2000, 408, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Murakami, A.; Tanaka, K. Skp1 stabilizes the conformation of F-box proteins. Biochem. Biophys. Res. Commun. 2011, 410, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G.; Elledge, S.J.; Conaway, R.C.; et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.S.; Skaar, J.R.; Pagano, M. SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem. Sci. 2012, 37, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xing, Y.; Yang, S.; Tian, D. Remarkable difference of somatic mutation patterns between oncogenes and tumor suppressor genes. Oncol. Rep. 2011, 26, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acis Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latres, E.; Chiarle, R.; Schulman, B.A.; Pavletich, N.P.; Pellicer, A.; Inghirami, G.; Pagano, M. Role of the F-box protein Skp2 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 2515–2520. [Google Scholar] [CrossRef] [Green Version]
- Von Der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; Nakayama, K.I.; et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar] [CrossRef]
- Ng, R.W.M.; Arooz, T.; Yam, C.H.; Chan, I.W.Y.; Lau, A.W.S.; Poon, R.Y.C. Characterization of the cullin and F-box protein partner Skp1. FEBS Lett. 1998, 438, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyctin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Siegfried, Z.; Simon, I. DNA methylation and gene expression. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 362–371. [Google Scholar] [CrossRef]
- Akhoondi, S.; Sun, D.; von der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Bretones, G.; Acosta, J.C.; Caraballo, J.M.; Ferrandiz, N.; Gomez-Casares, M.T.; Albajar, M.; Blanco, R.; Ruiz, P.; Hung, W.C.; Albero, M.P.; et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J. Biol. Chem. 2011, 286, 9815–9825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Du, L.; Ren, Y.; Liu, X.; Jiao, Q.; Cui, D.; Wen, M.; Wang, C.; Wei, G.; Wang, Y.; et al. SKP2 promotes breast cancer tumorigenesis and radiation tolerance through PDCD4 ubiquitination. J. Exp. Clin. Cancer Res. 2019, 38, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Ungermannova, D.; Chen, L.; Liu, X. A negatively charged amino acid in Skp2 is required for Skp2-Cks1 interaction and ubiquitination of p27Kip1. J. Biol. Chem. 2003, 278, 32390–32396. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Soengas, M.S.; Sun, Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res. 2009, 69, 4974–4982. [Google Scholar] [CrossRef] [Green Version]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef]
- Aziz, K.; Limzerwala, J.F.; Sturmlechner, I.; Hurley, E.; Zhang, C.; Jeganathan, K.B.; Nelson, G.; Bronk, S.; Fierro Velasco, R.O.; van Deursen, E.J.; et al. Ccne1 Overexpression Causes Chromosome Instability in Liver Cells and Liver Tumor Development in Mice. Gastroenterology 2019, 157, 210–226.e212. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Ecker, A.; Simma, O.; Hoelbl, A.; Kenner, L.; Beug, H.; Moriggl, R.; Sexl, V. The dark and the bright side of Stat3: Proto-oncogene and tumor-suppressor. Front. Biosci. 2009, 14, 2944–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [Green Version]
- Orlicky, S.; Tang, X.; Willems, A.; Tyers, M.; Sicheri, F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 2003, 112, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Postow, L.; Funabiki, H. An SCF complex containing Fbxl12 mediates DNA damage-induced Ku80 ubiquitylation. Cell Cycle 2013, 12, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Australian Ovarian Cancer Study, G.; Davis, S.; D’Andrea, A.D.; Simpson, K.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [Green Version]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A.; Komarova, N.L.; Sengupta, A.; Jallepalli, P.V.; Shih Ie, M.; Vogelstein, B.; Lengauer, C. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 2002, 99, 16226–16231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, R.; McManus, K.J. Chromosome Instability; Implications in Cancer Development, Progression, and Clinical Outcomes. Cancers 2020, 12, 824. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Su, Y.; Koeman, J.; Haak, E.; Dykema, K.; Essenberg, C.; Hudson, E.; Petillo, D.; Khoo, S.K.; Vande Woude, G.F. Chromosome instability drives phenotypic switching to metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 14793–14798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-to-Cell Heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.L.; McManus, K.J. A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS ONE 2015, 10, e0123200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Qiu, J.; Liu, Z.; Zeng, Y.; Fan, J.; Liu, Y.; Guo, Y. Overexpression of RING box protein-1 (RBX1) associated with poor prognosis of non-muscle-invasive bladder transitional cell carcinoma. J. Surg. Oncol. 2013, 107, 758–761. [Google Scholar] [CrossRef]
- Kunishige, T.; Migita, K.; Matsumoto, S.; Wakatsuki, K.; Nakade, H.; Miyao, S.; Kuniyasu, H.; Sho, M. Ring box protein-1 is associated with a poor prognosis and tumor progression in esophageal cancer. Oncol. Lett. 2020, 20, 2919–2927. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, Y.; Liu, J.; Sun, Y.; Jia, L. Protective autophagy induced by RBX1/ROC1 knockdown or CRL inactivation via modulating the DEPTOR-MTOR axis. Autophagy 2012, 8, 1856–1858. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Q.; Wang, X.L.; Cheng, X.; Lu, Y.Z.; Wang, G.Z.; Li, X.C.; Zhang, J.; Wen, Z.S.; Huang, Z.L.; Gao, Q.L.; et al. Skp1 in lung cancer: Clinical significance and therapeutic efficacy of its small molecule inhibitors. Oncotarget 2015, 6, 34953–34967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Lang, T.; Qiu, J.; Han, K.; Zhou, L.; Min, D.; Zhang, Z.; Qi, D. SKP1 promotes YAP-mediated colorectal cancer stemness via suppressing RASSF1. Cancer Cell. Int. 2020, 20, 579. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Xiong, D.B.; Huang, T.B.; Zheng, J.H.; Yao, X.D. Expression of CUL1 correlates with tumour-grade and recurrence in urothelial carcinoma. ANZ J. Surg. 2017, 87, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, G. Increased Cul1 expression promotes melanoma cell proliferation through regulating p27 expression. Int. J. Oncol. 2010, 37, 1339–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Zhou, Y.; Chen, G.; Zeng, J.; Ding, J.; Tan, Y.; Zhou, J.; Li, G. Overexpression of Cullin1 is associated with poor prognosis of patients with gastric cancer. Hum. Pathol. 2011, 42, 375–383. [Google Scholar] [CrossRef]
- Hershko, D.D. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008, 112, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw, M.D.; Liu, Y.; Zheng, Y.C.; Shi, X.J.; Liu, H.M. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med. Res. Rev. 2020, 40, 1920–1949. [Google Scholar] [CrossRef]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.C.; Pan, B.S.; Manne, R.; Li, H.Y.; Lin, H.K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef]
- Petersen, I.; Kotb, W.F.; Friedrich, K.H.; Schluns, K.; Bocking, A.; Dietel, M. Core classification of lung cancer: Correlating nuclear size and mitoses with ploidy and clinicopathological parameters. Lung Cancer 2009, 65, 312–318. [Google Scholar] [CrossRef]
- Zeimet, A.G.; Fiegl, H.; Goebel, G.; Kopp, F.; Allasia, C.; Reimer, D.; Steppan, I.; Mueller-Holzner, E.; Ehrlich, M.; Marth, C. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol. Oncol. 2011, 121, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galindo-Moreno, M.; Giraldez, S.; Limon-Mortes, M.C.; Belmonte-Fernandez, A.; Reed, S.I.; Saez, C.; Japon, M.A.; Tortolero, M.; Romero, F. SCF(FBXW7)-mediated degradation of p53 promotes cell recovery after UV-induced DNA damage. FASEB J. 2019, 33, 11420–11430. [Google Scholar] [CrossRef]
- Orr, B.; Compton, D.A. A double-edged sword: How oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front. Oncol. 2013, 3, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, S.; Cato, K.; Tang, L.; Pavey, S.; Haass, N.K.; Gabrielli, B.G.; Duijf, P.H. In vivo overexpression of Emi1 promotes chromosome instability and tumorigenesis. Oncogene 2016, 35, 5446–5455. [Google Scholar] [CrossRef]
- Erlanson, M.; Landberg, G. Prognostic implications of p27 and cyclin E protein contents in malignant lymphomas. Leuk. Lymphoma 2001, 40, 461–470. [Google Scholar] [CrossRef]
- Takada, M.; Zhang, W.; Suzuki, A.; Kuroda, T.S.; Yu, Z.; Inuzuka, H.; Gao, D.; Wan, L.; Zhuang, M.; Hu, L.; et al. FBW7 Loss Promotes Chromosomal Instability and Tumorigenesis via Cyclin E1/CDK2-Mediated Phosphorylation of CENP-A. Cancer Res. 2017, 77, 4881–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisyk, A.L.; Penner-Goeke, S.; Lichtensztejn, Z.; Nugent, Z.; Wightman, R.H.; Singh, H.; McManus, K.J. Characterizing the prevalence of chromosome instability in interval colorectal cancer. Neoplasia 2015, 17, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penner-Goeke, S.; Lichtensztejn, Z.; Neufeld, M.; Ali, J.L.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. The temporal dynamics of chromosome instability in ovarian cancer cell lines and primary patient samples. PLoS Genet. 2017, 13, e1006707. [Google Scholar] [CrossRef]
- Morden, C.R.; Farrell, A.C.; Sliwowski, M.; Lichtensztejn, Z.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. Chromosome instability is prevalent and dynamic in high-grade serous ovarian cancer patient samples. Gynecol. Oncol. 2021, 161, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, V.D.B. Ovarian Cancer Biomarkers: Moving Forward in Early Detection. Adv. Exp. Med. Biol. 2020, 1219, 355–363. [Google Scholar] [CrossRef]
- Moore, J.S.; Aulet, T.H. Colorectal Cancer Screening. Surg. Clin. N. Am. 2017, 97, 487–502. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Oehlmann, S.; Sowa, M.E.; Harper, J.W.; Pavletich, N.P. Structure of a Fbw7-Skp1-Cyclin E Complex: Multisite-Phosphorylated Substrate Recognition by SCF Ubiquitin Ligases. Mol. Cell 2007, 26, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, L.; Wahl, S.; Pechlivanis, S.; Hoffmann, P.; Schmid, M. A statistical model for the analysis of beta values in DNA methylation studies. BMC Bioinform. 2016, 17, 480. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos Gudiño, R.; Farrell, A.C.; Neudorf, N.M.; McManus, K.J. A Comprehensive Assessment of Genetic and Epigenetic Alterations Identifies Frequent Variations Impacting Six Prototypic SCF Complex Members. Int. J. Mol. Sci. 2022, 23, 84. https://doi.org/10.3390/ijms23010084
Campos Gudiño R, Farrell AC, Neudorf NM, McManus KJ. A Comprehensive Assessment of Genetic and Epigenetic Alterations Identifies Frequent Variations Impacting Six Prototypic SCF Complex Members. International Journal of Molecular Sciences. 2022; 23(1):84. https://doi.org/10.3390/ijms23010084
Chicago/Turabian StyleCampos Gudiño, Rubi, Ally C. Farrell, Nicole M. Neudorf, and Kirk J. McManus. 2022. "A Comprehensive Assessment of Genetic and Epigenetic Alterations Identifies Frequent Variations Impacting Six Prototypic SCF Complex Members" International Journal of Molecular Sciences 23, no. 1: 84. https://doi.org/10.3390/ijms23010084
APA StyleCampos Gudiño, R., Farrell, A. C., Neudorf, N. M., & McManus, K. J. (2022). A Comprehensive Assessment of Genetic and Epigenetic Alterations Identifies Frequent Variations Impacting Six Prototypic SCF Complex Members. International Journal of Molecular Sciences, 23(1), 84. https://doi.org/10.3390/ijms23010084