Abstract
Myeloid malignancy is a broad term encapsulating myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML). Initial studies into genomic profiles of these diseases have shown 2000 somatic mutations prevalent across the spectrum of myeloid blood disorders. Epigenetic mutations are emerging as critical components of disease progression, with mutations in genes controlling chromatin regulation and methylation/acetylation status. Genes such as DNA methyltransferase 3A (DNMT3A), ten eleven translocation methylcytosine dioxygenase 2 (TET2), additional sex combs-like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2) and isocitrate dehydrogenase 1/2 (IDH1/2) show functional impact in disease pathogenesis. In this review we discuss how current knowledge relating to disease progression, mutational profile and therapeutic potential is progressing and increasing understanding of myeloid malignancies.
1. Introduction
Haematopoiesis is a tightly regulated process involving gene expression control that guides conversion from progenitor cells to terminally differentiated haematopoietic cells. During leukaemogenesis, processes such as self-renewal, differentiation and cell expansion are disrupted, resulting in accumulation of immature blast cells. Initial cytogenetic studies in leukaemia identified chromosomal translocations which generate oncogenic fusion proteins mainly altering transcriptional regulators [1]. Subsequently, recurrent somatic mutations were discovered in genes encoding regulators of cellular differentiation and signalling that were required for malignant transformation. These classical models in leukaemia research together with the original two-hit hypothesis have been unsuccessful in explaining the distinctive characteristics of decreased differentiation in leukaemia [2]. Myeloid malignancies encapsulate a group of disorders caused by clonal expansion of haematopoietic stem/progenitor cells including acute myeloid leukaemia (AML), myelodysplastic syndrome (MDS) and myeloproliferative neoplasms (MPN) (polycythaemia vera, essential thrombocythemia and primary myelofibrosis). MPN and MDS possess the ability to transform into secondary AML.
The interplay of epigenetic processes, DNA methylation and histone modifications, determine if a (set of) gene(s) is transcriptionally expressed or silenced. Examples include methylation of CpG islands, (de)methylation, and (de)acetylation of histones. These processes regulate chromatin conformation and in turn regulate gene expression. Studies have revealed the essential role of deregulated epigenetic processes in AML [3,4]. Because epigenetic modifications are inherently reversible and dynamic, deregulated epigenetic mechanisms might provide a path towards targeted therapies via inhibitors of proteins that modify histones or DNA methylation. Chromatin structure has a large impact on the regulation of gene expression, and a lot more can be discovered about how individual epigenetic marks are set up and then maintained throughout DNA replication and cell division. Chemical modification of DNA or chromatin-associated proteins and histones have a major effect on chromatin structure and gene expression.
In this review, we take advantage of evidence from a range of somatic mutations frequently occurring in chromatin and epigenetic modifiers as well as explore the relationship between DNA methylation and histone modification. We will describe the relationship between these modifiers and leukaemogenic pathways leading to decreased cell differentiation and deviant self-renewal and proliferation. We then discuss how these data based on epigenetic modifiers have, and will continue to, lead to innovative epigenetic therapeutics. Finally, we address how understanding the combinatorial factors leading to AML will enable us to decipher epigenetic reprogramming and understand the mechanisms of gene regulation.
2. Current Knowledge of Genetic Profiles for Myeloid Malignancies
Mutations impacting myeloid malignancies can be divided by five classes: spliceosome components (e.g., Splicing factor 3B subunit 1 (SF3B1)), epigenetic regulators (e.g., DNA methyltransferase 3A (DNMT3A),), signalling pathway proteins (fms-like tyrosine kinase 3 (FLT3), Janus kinase 2 (JAK2)), transcription factors (CCAAT enhancer-binding protein alpha (CEBPA), runt-related transcription factor 1 (RUNX1)) and tumour suppressor proteins (Tumour protein p53 (TP53)) [5]. The last four decades have showed varying rates of development for clinical treatment for these myeloid disorders, encompassing chemotherapy and tyrosine kinase inhibitors (TKI’s) to more modern therapies which include epigenetically active inhibitors like histone deacetylase inhibitors (HDACi) and DNA methyltransferases (DNMT) that have an impact on chromatin structure and accessibility [6,7].
Multiple somatic mutations, which occur in genes encoding regulators of epigenetic modifiers, have shown to alter epigenetic and chromatin remodelling activity of the cell. Yet, the original two-hit hypothesis for myeloid progression does not cover all disease alleles. Mutant alleles that do not conform to either class have shown to promote epigenetic modifications and include alterations in DNMT3A and ten eleven translocation methylcytosine dioxygenase 2 (TET2). These data suggest there is an ever increasing need to explore the expanding biological and clinical relevance of epigenetic alterations in myeloid malignancies.
These epigenetic alterations influence a range of cellular processes having further impact outside of the epigenome through control of chromatin and gene expression. Indicating that chromatin and epigenetic modifiers embody a cluster of mutations essential for leukaemic development. It is widely accepted that epigenetic alterations rarely occur alone but culminate from a large profile of concomitant mutations (Figure 1).
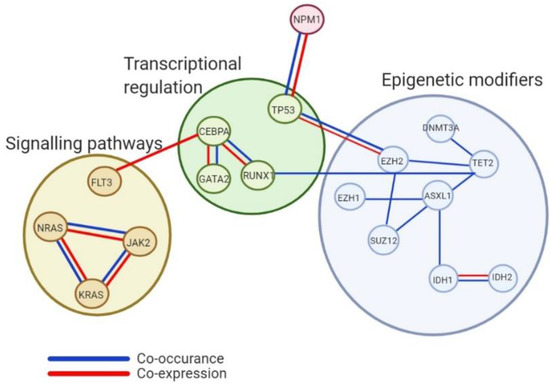
Figure 1.
Analysis of co-occurrence and co-expression of epigenetic genes and other cellular regulatory processes. Interactions shown between genes considered to possess epigenetic modification processes and other cellular processes like transcriptional regulation and signalling and kinase pathways highlighting the intricate pathway association needing accounted for in the progression of knowledge around mutational profiles within myeloid malignancies.
Recurrent cytogenetic events and somatic variants revealed from sequencing analyses uncovered 9 genetic classes off AML [5]. Deep sequencing studies across 200 patients by The Cancer Genome Atlas (TCGA) uncovered a heterogeneous disease with approximately 2,000 somatically mutated genes [8]. Majority of these recurrent cytogenetic events and somatic mutations play an important role in prognosis [5,9]. Some the most frequent somatic mutations have been detected in both MDS and MPN, as well as healthy individuals with age-related clonal haematopoiesis, thus associated with substantial risk for progression of myeloid malignancies [10,11]. The AML karyotype has been well documented in the European LeukemiaNet (ELN) risk stratification guide was revised in 2017 to outline the favourable-risk (FR), intermediate-risk (IR) and adverse-risk (AR) profiles. ‘Favourable-risk’ cytogenetic abnormalities include Acute Myeloid Leukaemia- Eight Twenty-Two (AML-ETO), also known as RUNX1 Translocation Partner 1 (RUNX1T1), Pro Myelocytic Leukaemia- Retinoic Acid Receptor alpha (PML-RARA) and Nucleophosmin 1 (NPM1) mutations [12,13]. Whereas most deletions (−5/del(5q)), inv(3)/t (3;3), fms-like tyrosine kinase 3- internal tandem duplications (FLT3-ITD) mutations and complex karyotypes are deemed as having an adverse prognostic outcome [1,14]. The abnormalities in intermediate-risk groups include cytogenetic abnormalities not listed in “favourable-risk” or adverse-risk groups or those with double mutations in CEBPA or no mutations in NPM/FLT3-ITD.
Functional investigations have highlighted two main classes of leukaemic disease alleles that contribute to the haematopoietic transformation targeting either signal transduction or epigenetic control in myeloid malignancies. Class I alleles, such as FLT3 and JAK2, have been shown to confer advantage to growth due to irregular activation and proliferation of cell signalling pathways (STAT, PI3K and RAS-MAPK) [15]. While class II alleles like mixed-lineage leukaemia (MLL) and CEBPα alter transcriptional targets used in haematopoietic differentiation and maturation [9,16].
3. Alterations in DNA Methylation
Methylation of DNA encompasses the addition of a methyl group to the 5-carbon position of cytosines in CpG dinucleotides producing 5-methylcytosine [17]. This is established by DNA methyltransferases (DNMTs). Altered DNA methylation patterns have been reported in the regions of CpG islands (gene promoters). The association of hypermethylation and hypomethylation of cytosines in CpG islands are pertinent in gene regulation. CpG islands are DNA methylation regions contained within promoters, they regulate expression of genes via transcriptional silencing of the corresponding gene [18].
Mutations occurring in DNMT3A is one of the most common mutations displayed in AML with ~20% of de novo patients displaying this aberration [19]. The DNMT3A gene encodes a methyltransferase that adds a methyl to the cytosine residue of CpG dinucleotides creating a C5 methylcytosine (5mC) (Figure 2A). Mutations in the DNMT3A gene were first identified and described by Ley et al. in 2010 after sequencing an AML patient who had a normal cytogenetic profile [19]. Increased methylation status was observed in DNMT3A mutants leading to reduced expression of downstream genes [20]. DNMT3A and its co-factor, DNMT3L, form an active tetramer with an increased affinity for DNA resulting in more efficient methylation (Figure 2B) [21]. Majority of mutations in the DNMT3A gene resulted in either nonsense/frameshift alterations which caused premature truncation of the protein product or an amino acid substitution at Arginine 882 (R882) (Figure 2A). In total, 65% of mutations in this amino acid are heterozygous missense mutations that exert a dominant effect over wild-type (WT) DNMT3A genes. R882 mutations have previously been shown to compromise methyltransferase activity resulting in increased self-renewal activity of haematopoietic stem cells (HSC) (Figure 2C) [22]. Koya et al. has shown that R882 mutants can dimerize with WT DNMT3A molecules but fail to form active tetramers resulting in a significant decrease in methyltransferase activity [23]. DNMT3 mutant R882 can interact with Polycomb Repressive complex 1 (PRC1) resulting in downregulation of haematopoietic differentiation genes like CEBPα, stunting cells in an immature state which still retain the capacity for self-renewal causing long-term expansion of HSC [24]. Crucial findings in highlighting the role of DNMT3AR882H mutations in AML chemoresistance showed the importance of aberrant recruitment of histone chaperone SPT-16 resulting in attenuated nucleosome eviction and chromatin remodelling as a result of exposure to anthracycline [25].
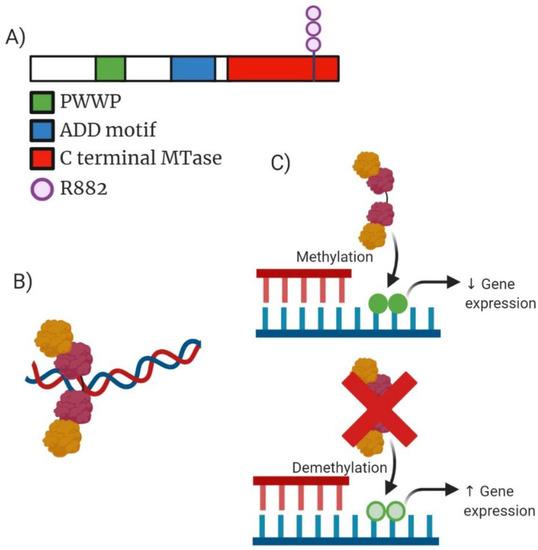
Figure 2.
Analysis of DNMT3A molecular structure, active tetramer structure, mechanism of action and drug target action. (A) Domain structure of mammalian DNMT3A enzyme consisting of 912 amino acid residues. Green highlighting PWWP domain which is required for directing DNA methylation. Blue highlighting ADD (ATRX-DNMT3A-DNMT3L) motif responsible for mediating protein–protein interaction to transcription factors. Red highlights the C-terminal MTase domain contains highly conserved regions of C5-DNA methyltransferases. Additionally, highlighted is the structural position of the most common mutation occurring within DNMT3A (R882). (B) Active tetramer formed from two DNMT3A and two DNMT3L molecules resulting in an increased affinity for DNA causing more efficient methylation. (C) Mechanism of action for both wild-type (WT) DNMT3A and the truncated mutant DNMT3A. WT DNMT3A causing methylation which can reduce expression of gene expression compared to mutant DNMT3A which can often result in increased expression of genes depending on the context of the cellular function occurring.
Many epigenetic alterations rarely occur alone in myeloid malignancies. Mutant DNMT3A is frequently associated with mutations in NPM1, FLT3 and isocitrate dehydrogenase 1/2 (IDH1/2), working together to cooperatively corrupt leukaemic progenitor cells. Patients who present with this collection of mutations tend to display a higher percentage of bone marrow blast cells while also experiencing a shorter time of event free survival (EFS) [26]. Patients with higher white blood counts (WBC) and platelets have been reported versus patients without the aberration. DNMT3A mutations display promiscuity in several myeloid malignancies and influence disease progression in MPS, MDS and de novo AML. Within AML patients it has been shown that DNMT3A mutations are enriched in those classified as M4/M5 AML by classification guidelines outlined by the French American-British classification system [27].
DNA methylation profiling provides information regarding diagnostic classification, prognostic stratification and therapeutic guidance in myeloid malignancies. Alterations in DNA methylation have been widely reported in most cancers, the result of these alterations promotes oncogenesis due to changes in gene expression [28]. Modern advancements in methylome analysis, e.g., microarray assays, have reduced time, cost and reproducibility of DNA methylation analysis, resulting in it becoming a reliable method in clinical diagnosis of myeloid malignancies [29,30,31]. It has been previously highlighted that known global methylation patterns assist in genetic characterisation of AML subgroups; Figueroa et al. reported distinct DNA methylation profiles which can be linked to characterisation of AML profiles indicating a clear link between methylation profiling and disease diagnosis leading to clinical decision making on treatment options [32,33].
4. Alterations in DNA Hydroxymethylation
The oxidation process of 5-methylcytosine to produce 5-hydroxymethylcytosine (5hmC) is catalysed by the TET-enzymes which is dependent on alpha-ketoglutarate (α-KG). TET2 was first discovered in 2009 with inactivating mutations occurring in MPM and AML patients, Delhommeau et al. accomplished this through mapping structural DNA rearrangements in chromosome 4q24 [34]. TET2 is a member of the TET family of proteins with mutations of this gene occurring in 15% of myeloid malignancies (Figure 3A). TET2-associated mutations have been observed in AML patients and are linked with reduced levels of 5hmC [35] as well as contribute to a poor prognosis in intermediate-risk AML [36,37].
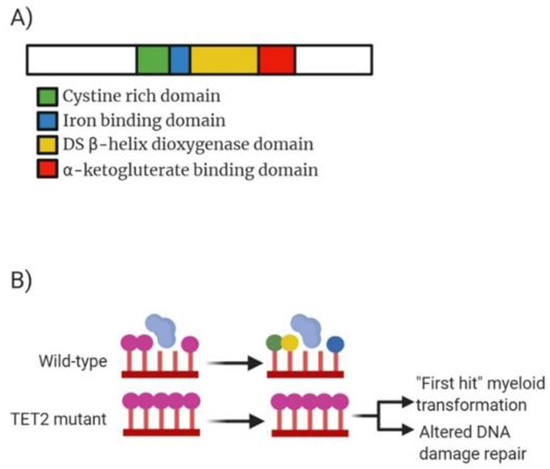
Figure 3.
Analysis of TET2 molecular structure, mechanism of action and drug target action. (A) Domain structure of mammalian TET2 enzyme consisting of 2002 amino acid residues. Green highlighting a cystine rich domain comprised of two sub-domains which modulate chromatin targeting by TET proteins. Blue highlighting the iron binding domain which interacts with double stranded β-helix domain (yellow) and α-ketoglutarate binding domain (red) to form a core catalytic region. DSBD also contains a low complexity insert whose function remains unclear. (B) Mechanism of action for both wild-type TET2 and the mutant TET2. WT TET2 causing conversion of 5-mC (purple circle) into 5-hmC (green), 5-fC (yellow) or 5-caC (blue). Mutated TET2 lowers expression resulting in overexpression of 5-hmC causing control over genes being switched on or off.
Genetic studies into TET2 have revealed the link between alterations in DNA methylation and tumour metabolism. Mutations in the widely recognised tumour suppressive TET2 gene have been shown to be sufficient to instigate myeloid malignancies in mice [38]. Mutations to the TET2 gene have been shown to include frameshift, early stop codons, deletions and AA substitutions highlighting a widely diverse profile for potential alterations. Majority of patients carry heterozygous TET2 mutations however a small proportion of patients present with a total homozygous loss of TET2. Previous work sequencing the coding exons of the gene by Jankowska et al. highlighted the association of homozygous mutations with uniparental disomy 4q compared to heterozygous mutations associated with lack of chromosomal lesions [39]. Studies have been reported on the mutational role of TET2 on progression of disease and suggested an involvement in early clonal mutations due to the presence of alterations in both myeloid and lymphoid progenitor compartments. Papaemmanuil et al. strengthened this by discovering a high allele frequency in TET2 mutations indicating that they occur as part of the “first hit” of mutational development in leukaemogenesis [5]. Once mutated, TET2 produces decreased levels of 5-hmC resulting in the unblocking of methyl-binding proteins (Figure 3B). Normal deposition of 5-hmC in CpG dinucleotides results in increased gene expression for those regions.
TET2 mutations have been observed to co-occur with NPM1, FLT3-ITD, JAK2, ASXL1, calreticulin (CALR), SF3B1 and RUNX1 revealing the range of epigenetic connections to cellular processes amongst various myeloid malignancies [40]. It is worth noting that mutations in TET2 are mutually exclusive together with Wilms’ tumour suppressor gene 1 (WT1), in 30–50% of AML patients. These patients present with a distinct AML subtype displaying dysregulated DNA hydroxymethylation. AML patients with TET2 mutations have a high WBC along with low platelet counts. Patients aged over 65 years old also experience low EFS (8.9 months vs. not reached) suggesting a role for TET2 as a prognostic biomarker [41]. AML patients defined as favourable risk (containing CEBPA and/or MPN1 without FLT3-ITD) by the European Leukaemia Net (ELN) will display worse EFS in the presence of TET2 mutations (10 months vs. 41.3 months) [41]. Yildirim et al. defined associations between 5-hmC and gene regulation through the interaction with methyl-CpG binding domain protein 3 (MBD3) a member of the NuRD complex (Nucleosome Remodelling and Deacetylase complex). They found that genes marked with 5-hmC were recognised by MBD3 and subsequently repressed [42]. The localisation of 5-hmC most often occurs in transcription sites and gene exons. The potential global impact of TET2 loss on the genome can lead to the prediction that it would lead to increased methylation and reduction of 5-hmC levels.
Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) proteins catalyse the conversion of isocitrate to α-KG. Mutations in genes encoding these IDH1/2 enzymes are frequently observed in AML patients with normal cytogenetics. IDH1/2 mutations result in aberrant metabolite 2-hydroxyglutarate (2-HG) [43] where it can compete with α-KG leading to TET2 inhibition. IDH1 mutations were discovered in 2008 by a sequencing project by Parsons et al. on human glioblastoma samples and since have been identified in AML patients by Mardis et al. [44,45]. IDH2 mutations were identified based on consequent genomic studies of IDH1 in AML [46]. Mutations in IDH proteins are found in 15–20% of AML cases [47]. Mutations in these genes are less common in MDS patients occurring in 3–5% of cases and 1–4% of MPN cases [48]. Mutational hotspots in IDH1 have shown to occur at Arg132 while IDH2 mutational hotspots occur at Arg140 more frequently than Arg172 [49] (Figure 4A). Ok et al. have shown that the persistence of IDH mutations lead to increased risk of relapse for patient’s post-treatment [50].
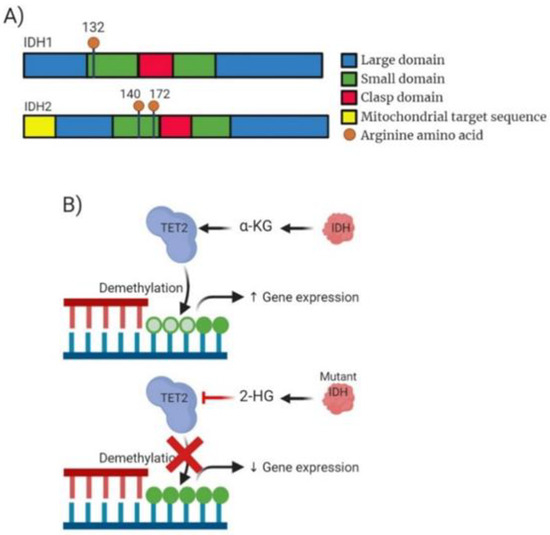
Figure 4.
Analysis of IDH molecular structure, mechanism of action and drug target action. (A) Domain structure of mammalian IDH1 enzyme consisting of 414 amino acid residues and IDH2 consisting of 452 amino acids. Blue highlighting a large domain. Green highlighting the small domain. Red highlighting clasp domain which links other subunits together. IDH2 also contains a mitochondrial targeting sequence. Additionally, highlighted is the structural position of the most common mutation occurring within IDH1 (R132) and IDH2 (R140/172). (B) Mechanism of action for both wild-type IDH and the mutant IDH. WT IDH causing production of α-KG enabling TET2 to function in demethylation of DNA causing potential increased gene expression. Mutated IDH inhibits TET2 function due to production of 2-HG expression often resulting decreased gene expression.
Patients presenting with IDH mutations are associated with distinct subsets of AML and normal/intermediate risk cytogenetic groups (27.1% of patients) suggesting that mutant IDH contribution can be linked to cell fate determination at the beginning stages of progenitor differentiation [26]. Clinical presentation includes older patients who present with higher platelet levels. Healthy individuals can also possess IDH mutations suggesting age-related clonal haematopoiesis; also suggesting the presence of IDH mutations in the beginning stages of leukaemogenesis and the requirement for concurrent mutations to enable AML progression. IDH mutations have been observed co-occurring alongside NPM1 mutations and have better prognosis [40]. IDH mutations have shown to be mutually exclusive with TET2 mutations. TET2, as described above, utilizes α-KG to enable the conversion of 5-mC into either 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5-fC) or 5-carboxylcytosine (5-caC) which is vital for the demethylation of DNA to control gene expression (Figure 4B). IDH mutations result in the levels of available α-KG being lowered in the cell due to the conversion to 2-HG. This action of IDH mutations exacerbates the impact of TET2 mutations resulting in greater levels of TET2-dependent demethylation of DNA. This indirect interaction between IDH and TET2 mutants results in increased global expression of 5-mC resulting in impaired DNA damage repair mechanisms or the promotion of myeloid malignancies.
5. Alterations in Histone/Lysine Methylation
The process of methylation on histone lysine residues can lead to mono-, di-, or trimethylation and is carried out by lysine methyltransferases (KMTs). These enzymes modify the affinity of reader proteins to methylated histones resulting in a context-dependent activation (H3K4, H3H36, and H3K79) or repression state (H3K9, H3K27, and H4K20) and whether mono-, di- or tri-methylation on the same lysine residue results in different functional consequences [51]. Post translational modifications (PTMs) are controlled by readers, writers and erasers which work in tandem to create, initiate or terminate biological signals within the cell [52]. The delicate balance of these activities controls the cellular regulation and any alterations in this balance can initiate dysregulation leading to human disease.
The lysine (K)-specific methyltransferase 2A (KMT2A) gene, also known as the mixed-lineage leukemia (MLL) gene, is a histone-lysine N-methyltransferase and leaves a mark of transcriptional activation by targeting H3K4. MLL is involved in translocations of approximately 10% of AML while mutations are frequently partial tandem duplications and found in 6% of de novo AML [53]. The normal MLL gene is located at 11q23 containing multiple motifs including AT hook DNA binding domains alongside domains for transcriptional activation and repression zinc finger domains [54]. MLL also contains a highly conserved SET domain which regulates homeotic (Hox) promoters [55]. The MLL gene contains an N terminal Menin binding domain. Menin protein acts a linker between the MLL gene and lens epithelium-derived growth factor (LEDGF), a chromatin binding protein [56]. Within the MLL gene there are 4 domains which are most commonly lost in the formation of MLL fusion proteins, these include the PHD finger, bromo-activation domain, activation domain and SET domain [57]. The majority of MLL rearrangements include the N terminal domain of the MLL gene and a C terminal domain of a translocation partner gene. Translocation partner genes can modulate transcription activity leading to activation of target genes or other diverse functions enabled by cytoplasmic/membrane bound partners [58]. There are more than 50 alternate translocation partners for the MLL gene that have been identified, each with unique biological properties. MLL has also been shown to interact with RUNX1 to regulate the epigenetic function of cis-regulatory haematopoietic genes. MLL translocations result in fusion proteins lacking the SET domain and replaced by members of the super elongation complex such as AF9 and AF10. This subset of translocated MLL partners account for ~90% of MLL recombinations [59]. A member subunit of this complex is DOT1L, a KMT that targets H3K79. AML samples with translocated MLL demonstrate deregulated H3K79 methylation, leading to the expression of target genes, in particular the homeobox genes [60].
EZH2 is a histone methyltransferase component of the Polycomb group (PcG) of proteins. EZH2 act as a transcriptional repressor and is a member of the Polycomb Repressive Complex 2 (PRC2). EZH2 contains a C-terminal SET domain which enables the protein to exert its methyltransferase activity (Figure 5A). EZH2, or its homologue EZH1, forms part of the canonical PRC2 along with embryonic ectoderm development (EED), suppressor of zeste 12 homologue (SUZ12) and RBBP4 (Figure 5B). EZH2 is unable to carry out this enzymatic function alone, the interaction with EED and SUZ12 enable gain of function. EZH2 functions to catalyse the addition of a methyl group to histone H3 at lysine 27 (H3K27) creating mono/dimethylation of H3K27 (H3K27me1/2) (Figure 5C) [61]. The methylation of H3K27 serves as an anchorage point for the recruitment of other PcG proteins contributing to gene repression impacting development and regulation of stem cell lineage development [61].
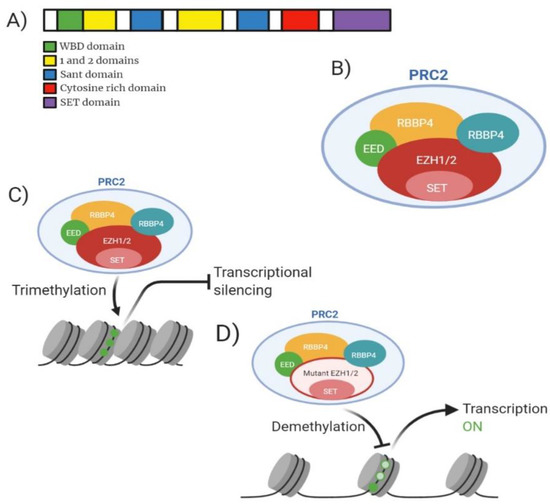
Figure 5.
Analysis of EZH2 molecular structure, PRC2 structure, mechanism of action for wild-type and mutant and drug target action. (A) Domain structure of mammalian EZH2 enzyme consisting of 751 amino acid residues. Green highlighting WDB (WD-40 binding domain). Yellow highlighting domain ‘1’ which binds PHF1 and domain ‘2’ binding region for SUZ12. Blue highlighting SANT domain allowing interaction of chromatin remodelling proteins with histones. Red highlights cytosine rich domain. Purple highlighting catalytic SET domain. (B) Model of mammalian PRC2 complex with core subunits. (C) Mechanism of action for wild-type EZH2 showing trimethylation of H3K27 resulting in gene transcription silencing. (D) Mechanism of action for PRC2 containing mutant EZH2 showing reduced methylation of H3K27 resulting in increased gene transcription.
EZH2 is implicated in 2.59% of MDS cases, 0.94% of AML cases and 0.80% of MPN cases and generally confer a poor prognosis [62,63]. EZH2 is the most frequently associated PRC2 member implicated in myeloid malignancies [9]. Majority of EZH2 heterozygous mutations occur at tyrosine 641 (Y641) located in the SET domain resulting in gain of function mutations [64]. It is also worth noting that studies suggested the dual role of EZH2 as an oncogene and a tumour suppressor. Tumour suppressor function of EZH2 was identified as an interaction partner with MLL-AF9/AML1-ETO9a AML [65]. Basheer et al. discovered that the deletion of EZH2 resulted in accelerated disease progression and reduced survival when occurring together with MLL-AF9/AML1-ETO9a suggesting that the presence of EZH2 can function to provide full oncogenic effects; adding another layer of complexity to the multitude of effects EZH2 contributes to cancer [65]. Mutations at Y641 can be a multitude of amino acid conversions; Y641F (Tyrosine-Phenylalanine), Y641N (Tyrosine-Asparagine), Y641S (Tyrosine-Serine), Y641H (Tyrosine-Histidine) or Y641C (Tyrosine-Cysteine). The mutants formed show increased affinity for H3K27me2 or H3K27me3 compared to WT-EZH2 which expresses preference for unmethylated H3K27 or H3K27me1 [66]. Shen et al. have shown that the knockout of EZH2 in embryonic stem cells reduces the methylation levels of H3K27me1/H3K27me2, however methylation is not removed in its entirety suggesting that EZH1 can contribute to the methylation status within the genome (Figure 5D) [67]. Patients presenting clinically with a myeloid malignancy may display lower blast percentage (21–30%) [68]. At diagnosis patients displaying mutant EZH2 may be slightly older than wild-type (59 years compared to 56 years) yet no differences are found between WBC counts, karyotype and ELN risk category of patients compared to wild-type [63].
ASXL1 is a human homologue of Drosophila additional sex combs gene and generally confers a poor prognosis, the gene is located on chromosome 20q11. [52]. Majority of the mutations that occur on the ASXL1 gene are at exon 12 and are heterozygous [53]. Structurally ASXL1 contains a conserved carboxyl-terminal PHD domain along with a HB1, ASXL1 restriction endonuclease helix-turn-helix (HARE-HTH) and a deubiquitinase adaptor (DEUBAD) (Figure 6A). ASXL1 is a member of a family of three identified genes (ASXL1, ASXL2 and ASXL3) which regulate chromatin remodelling but remain poorly characterised. ASXL1 functions within the PRC2 complex enabling cell differentiation via binding to the cohesion complex; controlling chromatin separation during cellular division. Scheuermann et al. highlighted the association between the polycomb-repressive deubiquitylase (PR-DUB) complex and its ability to deubiquitylate histone H2A at lysine 119 (H2AK119) (Figure 6B,C). This function can be accomplished by the components BRCA1-associated protein 1 (BAP1) and ASXL1 [54]. The ubiquitination of H2AK119 enables stabilization of PRC1/PRC2 enabling efficient deposition of H3K27me3 (Figure 6D).
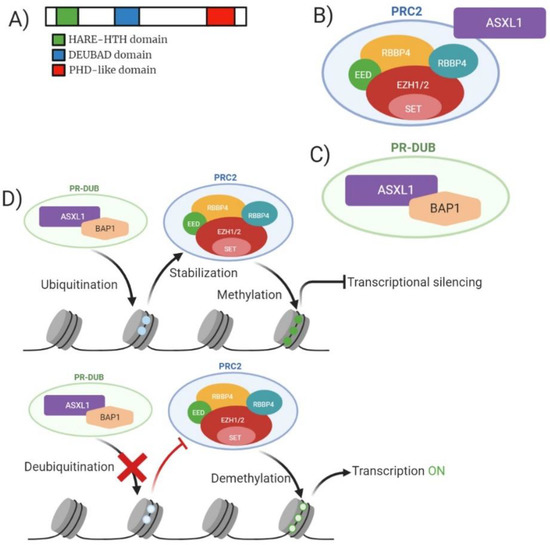
Figure 6.
Analysis of ASXL1 molecular structure, ASXL1/PRC2 structure, PR-DUB structure, and mechanism of action for wild-type and mutant ASXL1. (A) Domain structure of mammalian ASXL1 enzyme consisting of 1541 amino acid residues. Green highlighting HARE-HTH (HB1, ASXL1 restriction endonuclease helix-turn-helix). Yellow highlighting DEUBAD (deubiquitinase adaptor). Blue highlighting c terminal PHD domain. (B) Model of mammalian PRC2 complex with core subunits and association with ASXL1. (C) Model of PR-DUB complex with BAP1 and ASXL1. (D) Mechanism of action for both wild-type and mutant ASXL1 in relation to function with PRC2. Wild-type ASXL1 contributes to ubiquitination of H2AK119 enabling stabilization of PRC2 leading to trimethylation of H3K27 resulting in transcriptional silencing. Mutant ASXL1 causes deubiquitylation of H2SK119 preventing the stabilization of PRC2 resulting in reduced methylation of H3K27 resulting in increased gene transcription.
ASXL1 is shown to be implicated in 11–29% of MPN and MDS cases, and 17% of AML patients. Genetic studies into the mutational profile of ASXL1 suggest a higher occurrence of frameshift and nonsense mutations hence resulting in premature truncation of the protein upstream of the PHD finger. Truncation of this protein alters the functional state of the ASXL1 protein and the association it holds with PRC2 preventing chromatin remodelling from being controlled- this also suggests a highlighted role of ASXL1 being essential for roles of PRC1/2 (Figure 6D). In total, 50% of mutations in ASXL1 pertain to the duplication of a guanine nucleotide (c.1924drpG) resulting in a frameshift (p.Gly646TrpfsX12) [69]. The loss or mutation of ASXL1 alters the targets of the proteins including HOXA gene cluster. Expression of these gene clusters is directly increased by ASXL1 loss resulting in poorer prognosis for AML due to increased inappropriate expression of genes implicated in differentiation [70,71]. Clinical presentation of patients with ASXL1 are typically older male patients presenting with a more immature phenotype. There is a decrease in bone marrow and circulating blasts in these patients. Patients with mutated ASXL1 experience lower complete remission (CR) than WT ASXL1 patients (56% vs. 74%), with a 5-year EFS (15.9% vs. 29%). Patients with ASXL1 are often associated with intermediate risk cytogenetics, and in this circumstance further association with trisomy 8 and del(7q)/-7 [72].
Similar to the previous mutations mentioned above mutated ASXL1 co-occurs frequently with IDH2R140 and RUNX1 mutations while being rarely detected with NPM1, FLT3-ITD or DNTM3A. ASXL1 is related in function to PRC2 but there is no evidence supporting mutual exclusivity with members of the PRC2 complex. It has also been proposed that ASXL1 can occur concomitantly with mutations in encoding signals (JAK2, RAS) and splicing proteins (SF3B1) [69]. ASXL1 interacts with binding partner BAP1 however there is no evidence supporting mutations of BAP1 being implicated in myeloid malignancies [69]. Patients possessing both ASXL1 and RUNX1 experience a poorer prognostic outcome. It has been suggested by Bera et al. that cooperation of ASXL1 and RUNX1 play a critical role in leukaemic transformation [73]. Patients with these cooperating mutations experience amplified proliferation of mutated cells with a progression block of differentiation and increased self-renewal. Mice transduced with these mutations heightened inhibition of DNA binding 1 protein inhibitor (ID1) expression. ID1 is a transcriptional regulator that negatively regulates basic helix-loop-helix transcription factors inhibiting DNA binding and controlling transcriptional activity.
6. Targeted Epigenetic Therapies
Epigenetic alterations are an emerging field of cancer research and as such therapeutic options are limited. Hypomethylating agents are used as DNA methyltransferase inhibitors. Agents such as azacytidine and decitabine have shown success in the treatment of MDS [74]. Decitabine is a deoxycytidine analogue which can be incorporated into DNA during the S phase of replication. This results in decitabine binding to the DNA methyltransferase which obstructs the actions rendering it inactive. In single agent trials decitabine achieved complete remission (CR) rate of 24% with a median overall survival of 7.7 months for elderly patients unable to complete intensive standard care chemotherapy [75]. Azacytidine is a cytidine analogue primarily incorporated into RNA acting to inhibit RNA processing and functions. Azacytidine holds the ability to be converted to decitabine to exert its effects onto DNA also. As a single agent used to treat leukaemic malignancies azacytidine showed an OS of 12.1 months alongside a CR of 46.5% [76]. These agents have been shown to cause a reduction in global methylation marks in the genome creating a synthetically lethal phenotype leading to targeted cell death. DNMT3A has been shown to interact with and upregulate the HOX (homeobox) family of proteins. HOX proteins encode master regulators of embryonic development [77]. A range of agents which target HOX cluster genes; including DOTL1 (EPZ-5676), MLL-menin interaction inhibitors and MLL inhibitors all of which showed knockdown of the cluster genes resulting in leukaemic cell death [78,79]. The use of these inhibitors shows how targeting biological processes impacted by the mutation can result in lessening of prognostic outcome on the patient. Growing knowledge of the interactions between epigenetic modifications suggest single agent therapies may not be adequate alone. Combination therapies have been the focus of many studies as the knowledge of epigenetics deepens. One such combination that showed promising results was azacytidine alongside Trichostatin A (TSA; HDAC inhibitor). Using these therapies together showed synergistic actions with the display of decreased cell viability and tumorigenic ability alongside an increase in anti-oncogenic expression of cells. In single agent experiments TSA showed no activity against methylated genes while azacytidine showed only weak induction of transcription [80]. A phase 2 clinical trial by Garcia-Manero et al. showed studies using azacytidine alongside a HDACi yielded a lower overall survival (OS) (19.1 months) and complete remission (CR) (52%) compared to monotherapy with azacytidine [81]. There are multiple active clinical trials currently combining these hypomethylating drugs with other compounds to assess combined impact. Trial NCT01869114 has combined Azacytidine with Sirolimus, an immunosuppressant commonly used to prevent kidney rejection, to assess the impact on MDS and AML patients unable to tolerate high dose chemotherapy (ClinicalTrials.gov identifier NCT01869114). Decitibine is currently being trialled alongside Ruxolitinib, a drug commonly used in the treatment of myelofibrosis, to assess the efficacy of an intensified conditioning regimen in patients considered high risk within haematological malignancies currently undergoing SC transplants (ClinicalTrials.gov identifier NCT04582604).
TET2 can be used as a predictor for treatment response for MDS patients. Bejar et al. examined mutations for their association with overall survival. They found patients presenting with TET2, but lacking clonal ASXL1 mutations, showed highest response rates (OS = 3.65). It was also shown that response to Azacytidine treatment post bone marrow transplant was increased in patients who lacked TET2 [82]. It can be theorised that the lack of ASXL1 removed the potential resistance to hypomethylating agents (HMAs) that can be conferred. While mutant TET2s mechanism of action upon sensitivity to azacytidine is still unclear. Some myeloid malignancies have been shown to display resistance to HMAs providing issues for patient treatment. TET2 has been shown to act as a scaffolding protein to recruit components for DNA damage repair enabling another mechanistic avenue to exploit. PARP (poly ADP-ribose polymerase) inhibitors (PARPi) have provided an alternative avenue for potentially targeting these HMA-resistance neoplasms. It has been shown that TET2 mutation results in downregulation of BRCA2- which can cause sensitisation to PARPi, TET2 reduction has also been shown to impact upon homologous recombination (HR) [83]. This combination of cellular alterations means that the impairment of DNA damage along with treatment of DNA damaging chemotherapeutic agents may increase sensitivity to PARPi leading to better patient treatment for TET2 mutant individuals [84]. A recent 2019 study conducted by Das et al. investigated the use of ascorbate (Vitamin C) treatment for TET2 mutant AML patients [85]. Ascorbate is required, along with 2-oxygluterate, iron and oxygen for the function of TET dioxygenases enabling the active demethylation of DNA. It has been shown ascorbate deficiency mimics the impact of TET2 loss by interacting with FLT3 resulting in leukaemogenesis- ascorbate replenishment has been suggested to reverse these changes due to increasing TET2 levels [85,86]. The action of high dose ascorbate treatment promoted DNA demethylation inducing stem cell maturation while suppression of leukaemic cells providing a potential alternative treatment options for AML patients [87]. Current clinical trials include NCT03682029 which investigates the role of oral vitamin C administration to potentially change the biology of low-risk myeloid malignancies (ClinicalTrials.gov identifier NCT03682029).
The use of hypomethylating agents against IDH mutations was assessed by DiNardo et al. [88]. When treated with decitabine and azacytidine as single therapies or in combination with current front-line therapy; there was no association found between IDH status and treatment response. The FDA approved Ivosidenib (AG-120) and Enasidenib (AG-221) target mutant IDH1 and IDH2, respectively by blocking the production of 2-HG leading to reduced blast count and an increase in mature myeloid cells. A number of small molecule inhibitors are being developed to target IDH mutational products. This range of inhibitors are being directed against the hypermethylation of histones and DNA in cells containing mutant IDH. The over expression of 2-HG can also be targeted with AGI-5198 (selective inhibitor targeting IDH1-R132) by blocking the production of 2-HG by mutant cells resulting in the restoration of differentiation and proliferation in leukaemic cells [89]. Another inhibitor HMS-101 targeting IDH1 has been developed and showed to induce cell death within bone marrow cells expressing mutant IDH1 decreasing the mutant colony population [90]. The products of mutant IDH can be indirectly inhibited, where studies have suggested that glutaminase, an enzyme responsible for producing glutamine, can inhibit the growth of IDH mutant AML cells [91]. The inhibition of glutaminase alters the ability for cells to provide a production source for α-KG and 2-HG.
EZH2 mutants showed concomitant occurrence with RUNX1, ASXL1, NRAS and NPM1 suggesting that these mutations are associated with secondary AML. Deletion of the EZH2 gene from cells resulted in aberrant cell cycle entry along with an increase in apoptotic cell death which promoted a potential therapeutic target for patients with this mutation [92]. Inhibition of EZH2 is an emerging field in terms of myeloid therapy. Xu et al. investigated the role of UNC1999 (a dual inhibitor targeting both EZH1 and EZH2) showing anti-leukaemic effects against MLL-ENL cells resulting in differentiation of general cell population and apoptosis of mutated cells [93]. More recently Scheepstra et al. highlighted the potential to optimise proteolysis-targeting chimera (PROTAC) mediated protein degradation has displayed anti-tumour effects against epigenetic regulators, e.g., TRIM24 suggesting potential application to EZH2 methyltransferase activity, which requires further work into understanding the mechanism of action for EZH2 [94]. Swords et al. presented evidence supporting the interaction between EZH2 and retinoic acid in AML patients [95]. They showed that EZH2 mutations can impede the differentiation process by blocking retinoic acid (RA) differentiation due to the regulation of RA signalling by PRC2. This suggests the potential impact of epigenetic reprogramming (via inhibition by epigenetic inhibitors, i.e., EZH2i) sensitising cells to all-trans retinoic acid (ATRA) to produce a cumulative therapeutic result. There is evidence suggesting the combination of EZH2 inhibitors (i.e., S-adenosylhomocysteine hydrolase inhibitor 3-deazaneplanocin A (DZNep)) and Panobinostat (histone deacetylase inhibitor) results in apoptosis of AML progenitor cells by inhibition of H3K27me3 along with deacetylation of histones [96]. Tazemetostat is a first in class oral EZH2 inhibitor currently used in multiple clinical trials for patients with follicular lymphoma (ClinicalTrials.gov identifier NCT03456726 and NCT01897571) [97]. Information on all therapies mentioned are summarized within Table 1.

Table 1.
Table showing overview of mentioned drugs currently in development or approved for treatment of myeloid malignancies via epigenetic control.
Therapeutic treatment targeting ASXL1 mutations are limited, however patients with ASXL1 and RUNX1 mutations show poor response to HMAs hence causing reduced survival [98].
7. Future Perspectives, Prospects, Directions and Conclusion
The importance of the role of deregulated epigenetics in myeloid malignancies is rapidly evolving enabling improved patient stratification and treatment. A range of epigenetic modifiers regulating methylation of DNA (i.e., DNMT3A, IDH1, IDH1, TET2) or modification of histones (EZH2 and ASXL1) provide a targetable mutation for a subset of patients which enhancing knowledge around concomitant expression of mutations in myeloid malignancies (Table 2). The highlighted effects of these mutations on methylation and histone alterations suggests further investigation into the response of the epigenome when treated with inhibitors for MPN, MDS and AML patients. Mutations in epigenetics can be associated with prognostic outcome with DNMT3A and ASXL1 being associated with poorer outcomes highlighting the need for novel therapies targeting these high-risk aberrations. A number of these mutations remain vague in their functions and biological consequences for patients. The global impact upon the genome of these mutations needs to be evaluated further to understand how best to treat patients. Alterations in global chromatin architecture contribute to progression of myeloid malignancies and the interaction of concurrent mutations contribute to patient’s pathogenesis while fully elucidating precise interactions between proteins implicated. Recent advancements in the understanding of mutations in the (epi)genome have paved the way for epigenome exploration.
Table 2.
Table summarizing the epigenetic modifiers including frequency within disease, biological activity, and concomitant mutations. R882- Arginine 882. 5-mC- 5-methylcytosine. 5-hmC- 5-hydroxymethylcytosine. 5-fC- 5-formylcytosine. F-caC- 5-carboxylcytosine (5-caC). α-KG- α-ketoglutarate. 2-HG- 2-hydroxyglutarate. PRC2- Polycomb repressive complex 2. H3K27- Histone 3 Lysine 27.
Author Contributions
Writing—original draft preparation, writing—review and editing, D.V.; supervision, K.IM. and A.M.-S.; funding acquisition, Leukaemia and Lymphoma NI. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Leukaemia and Lymphoma NI, grant numbers R2536CNR and R2460CNR.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K.; on behalf of the National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Morison, J.; Dawson, M.A. Epigenetics in the hematologic malignancies [Internet]. Haematologica 2014, 997, 1772–1783. Available online: http://www.haematologica.org/content/99/12/1772 (accessed on 7 June 2020). [CrossRef] [PubMed]
- Abdel-Wahab, O.; Levine, R.L. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013, 121, 3563–3572. Available online: https://pubmed.ncbi.nlm.nih.gov/23640996/ (accessed on 11 June 2020). [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug therapy for acute myeloid leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef]
- Claus, R.; Rüter, B.; Lübbert, M. Targets of epigenetic therapy—Gene reactivation as a novel approach in MDS treatment. Cancer Treat. Rev. 2007, 33 (Suppl. 1), S47–S52. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2015, 263, 22–35. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Deininger, M.W.; Tyner, J.W.; Solary, E. Turning the tide in myelodysplastic/myeloproliferative neoplasms. Nat. Rev. 2017, 17, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Preudhomme, C.; Lai, J.L.; Morel, P.; Beuscart, R.; Bauters, F. Cytogenetics and their prognostic value in de novo acute myeloid leukaemia: A report on 283 cases. Br. J. Haematol. 1989, 73, 61–67. [Google Scholar] [CrossRef]
- Schoch, C.; Haferlach, T.; Haase, D.; Fonatsch, C.; Löffler, H.; Schlegelberger, B.; Staib, P.; Sauerland, M.C.; Heinecke, A.; Büchner, T.; et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: A study of 90 patients. Br. J. Haematol. 2001, 112, 118–126. [Google Scholar] [CrossRef]
- Haferlach, T.; Kern, W.; Schoch, C.; Schnittger, S.; Sauerland, M.C.; Heinecke, A.; Büchner, T.; Hiddemann, W. A new prognostic score for patients with acute myeloid leukemia based on cytogenetics and early blast clearance in trials of the German AML Cooperative Group. Hematology 2004, 89, 408–418. [Google Scholar]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute myeloid leukemia—genetic alterations and their clinical prognosis. Int. J. Hematol. Stem Cell Res. 2017, 11, 329–339. [Google Scholar]
- Di Nardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA methylation in acute myeloid leukemia and its clinical implications. Int. J. Mol. Sci. 2019, 20, 4576. [Google Scholar] [CrossRef]
- Lim, W.-J.; Kim, K.H.; Kim, J.-Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated With Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.L.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.J.; et al. DNMT3AMutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816.e13. [Google Scholar] [CrossRef]
- Duymich, C.E.; Charlet, J.; Yang, X.; Jones, P.A.; Liang, G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 2016, 7, 11453. [Google Scholar] [CrossRef]
- Yuan, X.Q.; Peng, L.; Zeng, W.J.; Jiang, B.Y.; Li, G.C.; Chen, X.P. DNMT3A r882 mutations predict a poor prognosis in AML a meta-analysis from 4474 patients. Medicine 2016, 95, 3519. [Google Scholar] [CrossRef]
- Koya, J.; Kataoka, K.; Sato, T.; Bando, M.; Kato, Y.; Tsuruta-Kishino, T.; Kobayashi, H.; Narukawa, K.; Miyoshi, H.; Shirahige, K.; et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat. Commun. 2016, 7, 10924. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a030320. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Loghavi, S.; Zuo, Z.; Ravandi, F.; Kantarjian, H.M.; Bueso-Ramos, C.; Zhang, L.; Singh, R.R.; Patel, K.P.; Medeiros, L.J.; Stingo, F.; et al. Clinical features of De Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J. Hematol. Oncol. 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Thol, F.; Damm, F.; Lüdeking, A.; Winschel, C.; Wagner, K.; Morgan, M.; Yun, H.; Göhring, G.; Schlegelberger, B.; Hoelzer, D.; et al. Incidence and Prognostic Influence of DNMT3A Mutations in Acute Myeloid Leukemia. J. Clin. Oncol. 2011, 29, 2889–2896. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. Available online: https://www.nature.com/articles/ng1089z (accessed on 13 July 2020). [CrossRef]
- Bock, C.; Halbritter, F.; Carmona, F.J.; Tierling, S.; Datlinger, P.; Assenov, Y.; Berdasco, M.; Bergmann, A.K.; Booher, K.; Busato, F.; et al. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726. [Google Scholar] [CrossRef]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef]
- Allum, F.; Shao, X.; Guénard, F.; Simon, M.M.; Busche, S.; Caron, M.; Lambourne, J.; Lessard, J.; Tandre, K.; Hedman, Å.K.; et al. Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants. Nat. Commun. 2015, 6, 1–12. Available online: www.nature.com/naturecommunications (accessed on 15 March 2021).
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Vosberg, S.; Kerbs, P.; Jurinovic, V.; Metzeler, K.H.; Amler, S.; Sauerland, C.; Görlich, D.; Berdel, W.E.; Braess, J.; Hiddemann, W.; et al. DNA Methylation Profiling of AML Reveals Epigenetic Subgroups with Distinct Clinical Outcome. Blood 2019, 134, 2715. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Becker, H.; Curfman, J.; Holland, K.B.; Schwind, S.; Whitman, S.P.; et al. TET2 Mutations Improve the New European LeukemiaNet Risk Classification of Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2011, 29, 1373–1381. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nat. Cell Biol. 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.-L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.-C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Kern, W.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD Complex Regulates Expression of 5-Hydroxymethylcytosine Marked Genes in Embryonic Stem Cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.-Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1andIDH2Gene Mutations Identify Novel Molecular Subsets Within De Novo Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Kattih, B.; Shirvani, A.; Klement, P.; Garrido, A.M.; Gabdoulline, R.; Liebich, A.; Brandes, M.; Chaturvedi, A.; Seeger, T.; Thol, F.; et al. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction—a retrospective propensity score analysis. Leukemia 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yonal-Hindilerden, I.; Daglar-Aday, A.; Hindilerden, F.; Akadam-Teker, B.; Yilmaz, C.; Nalcaci, M.; Yavuz, A.S.; Sargin, D. The Clinical Significance of IDH Mutations in Essential Thrombocythemia and Primary Myelofibrosis. J. Clin. Med. Res. 2016, 8, 29–39. [Google Scholar] [CrossRef]
- Im, A.P.; Sehgal, A.R.; Carroll, M.P.; Smith, B.D.; Tefferi, A.; Johnson, D.E.; Boyiadzis, M. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: Associations with prognosis and potential treatment strategies. Leukemia 2014, 28, 1774–1783. [Google Scholar] [CrossRef]
- Ok, C.Y.; Loghavi, S.; Sui, D.; Wei, P.; Kanagal-Shamanna, R.; Yin, C.C.; Zuo, Z.; Routbort, M.J.; Tang, G.; Tang, Z.; et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica 2019, 104, 305–311. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Klein, B.J.; Krajewski, K.; Restrepo, S.; Lewis, P.W.; Strahl, B.D.; Kutateladze, T.G. Recognition of cancer mutations in histone H3K36 by epigenetic writers and readers. Epigenetics 2018, 13, 683–692. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development [Internet]. Nat. Rev. Cancer 2007, 7, 823–833. Available online: www.nature.com/reviews/cancer (accessed on 29 June 2020). [CrossRef]
- Munoz, L.; Nomdedeu, J.F.; Villamor, N.; Guardia, R.; Colomer, D.; Ribera, J.M.; Torres, J.P.; Berlanga, J.J.; Fernandez, C.; Llorente, A.; et al. Acute myeloid leukemia with MLL rearrangements: Clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia 2003, 17, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Margolin, J.; Poplack, D.G.; Rabin, K.R. Molecular Genetics of Acute Lymphoblastic Leukemia. In The Molecular Basis of Cancer: Fourth Edition; Elsevier BV: Amsterdam, The Netherlands, 2015; pp. 395–406.e1. [Google Scholar]
- Winters, A.C.; Bernt, K.M. MLL-rearranged leukemias- An update on science and clinical approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Zeleznik-Le, N.J.; Harden, A.M.; Rowley, J.D. 11q23 translocations split the "AT-hook" cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. USA 1994, 91, 10610–10614. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.A.; Rubnitz, J.E. Acute Myeloid Leukemia in Children; Elsevier BV: Amsterdam, The Netherlands, 2018; pp. 981–993. [Google Scholar]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. Targeting Epigenetic Programs in MLL-Rearranged Leukemias. Hematology 2011, 2011, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Q.; Zi, J.; Ma, J.; Song, H.; Tian, Y.; McGrath, M.; Song, C.; Ge, Z. Mutations in EZH2 are associated with poor prognosis for patients with myeloid neoplasms. Genes Dis. 2019, 6, 276–281. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Rollig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 Mutations and Impact on Clinical Outcome Analyzed in 1604 Patients with Acute Myeloid Leukemia. Blood 2018, 132, 1528. [Google Scholar] [CrossRef]
- Kirmizis, A.; Bartley, S.M.; Kuzmichev, A.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004, 18, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Basheer, F.; Giotopoulos, G.; Meduri, E.; Yun, H.; Mazan, M.; Sasca, D.; Gallipoli, P.; Marando, L.; Gozdecka, M.; Asby, R.; et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019, 216, 966–981. [Google Scholar] [CrossRef]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.-W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Hsu, Y.-J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.-C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dai, H.; Wang, Q.; Wang, Q.; Xu, Y.; Wang, Y.; Sun, A.; Ruan, J.; Chen, S.; Wu, D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. PLoS ONE 2013, 8, e61341. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; DeVillier, R.; Murati, A.; Mozziconacci, M.-J.; Birnbaum, D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef]
- Chen, S.-L.; Qin, Z.-Y.; Hu, F.; Wang, Y.; Dai, Y.-J.; Liang, Y. The Role of the HOXA Gene Family in Acute Myeloid Leukemia. Genes 2019, 10, 621. [Google Scholar] [CrossRef]
- Pratcorona, M.; Abbas, S.; Sanders, M.A.; Koenders, J.E.; Kavelaars, F.G.; Erpelinck-Verschueren, C.A.; Zeilemakers, A.; Löwenberg, B.; Valk, P.J. Acquired mutations in ASXL1 in acute myeloid leukemia: Prevalence and prognostic value. Hematology 2011, 97, 388–392. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Herzig, J.K.; Aulitzky, T.; Bullinger, L.; Späth, D.; Teleanu, V.; Kündgen, A.; Köhne, C.-H.; et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: A study by the German-Austrian Acute Myeloid Leukemia Study Group. Hematology 2015, 100, 324–330. [Google Scholar] [CrossRef]
- Bera, R.; Chiu, M.-C.; Huang, Y.-J.; Lin, T.-H.; Kuo, M.-C.; Shih, L.-Y. RUNX1 mutations promote leukemogenesis of myeloid malignancies in ASXL1-mutated leukemia. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Paul, T.A.; Bies, J.; Small, N.; Wolff, L. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood 2010, 115, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Cashen, A.F.; Schiller, G.J.; O’Donnell, M.R.; DiPersio, J.F. Multicenter, Phase II Study of Decitabine for the First-Line Treatment of Older Patients With Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.D.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Lappin, T.R.; Grier, D.G.; Thompson, A.; Halliday, H.L. HOX genes: Seductive science, mysterious mechanisms. Ulst. Med. J. 2006, 75, 23–31. [Google Scholar]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia invivo. Cancer Cell 2015, 27, 589–602. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25817203 (accessed on 14 July 2020). [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Abaza, Y.; Takahashi, K.; Medeiros, B.C.; Arellano, M.; Khaled, S.K.; Patnaik, M.; Odenike, O.; Sayar, H.; Tummala, M.; et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: Results of a phase 2 study. Blood Adv. 2019, 3, 508–518. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Rashidi, A.; Amarillo, I.; Fisher, S.I. BRCA2-associated therapy-related acute myeloid leukemia. Med. Oncol. 2014, 32, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef]
- Das, A.B.; Kakadia, P.M.; Wojcik, D.; Pemberton, L.; Browett, P.J.; Bohlander, S.K.; Vissers, M.C.M. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J. 2019, 9, 1–5. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [PubMed]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome with Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Patel, K.P.; Garcia-Manero, G.; Luthra, R.; Pierce, S.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pemmaraju, N.; Konopleva, M.; et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk. Lymphoma 2014, 55, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.-A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. (R)-2-Hydroxyglutarate Is Sufficient to Promote Leukemogenesis and Its Effects Are Reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Cruz, M.M.A.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Li, B.; Chng, W.-J. EZH2 abnormalities in lymphoid malignancies: Underlying mechanisms and therapeutic implications. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125, 346–357. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.; van Hijfte, L.; Folmer, R.H. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Swords, R.T.; Perez, A.; Rodriguez, A.; Watts, J.M.; Schenk, T.; Vargas, F.; Elias, R.; Zelent, A. In Acute Myeloid Leukemia (AML), Targeting the Histone Methyltransferase EZH2 Promotes Differentiation, Impairs Clonogenic Survival and Augments the Anti-Leukemic Effects of the Retinoid, All-Trans-Retinoic Acid (ATRA). Blood 2015, 126, 3786. [Google Scholar] [CrossRef]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Weng, J.; Li, M.; Lu, Z.; Deng, C.; Sun, Q.; Xu, R.; Geng, S.; Du, X. Co-occurrence of RUNX1 and ASXL1 mutations underlie poor response and outcome for MDS patients treated with HMAs. Am. J. Transl. Res. 2019, 11, 3651–3658. [Google Scholar] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerová, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).