
Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models

 , , , , and
, , , , and 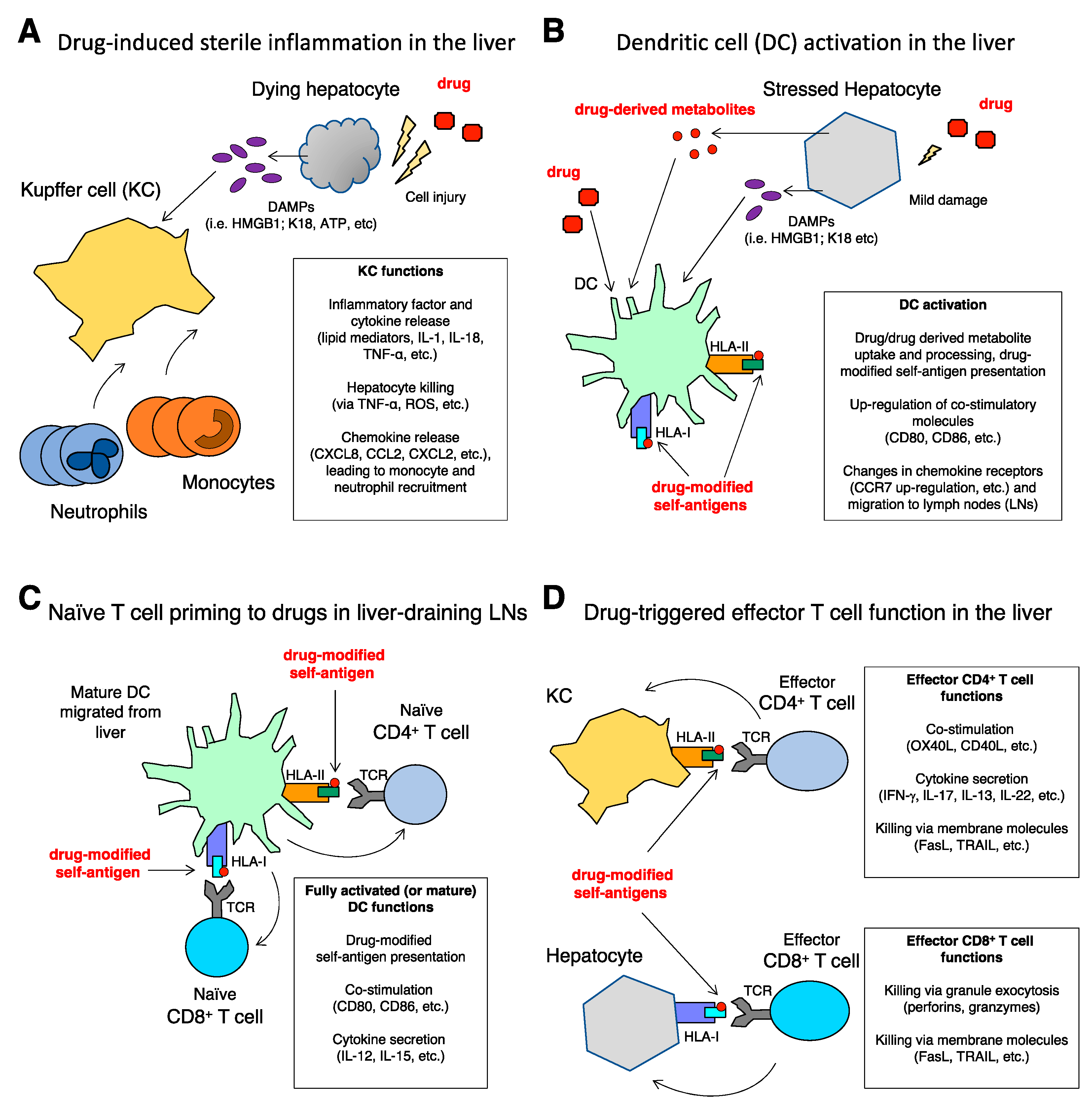{kind=link}
Abstract
1. Introduction
1.1. Types of DILI
1.2. DILI vs. AIH: A Clinical Challenge
2. Genetics of DILI
3. Immunology of DILI
3.1. Intrinsic DILI: Amplification of Liver Damage by Inflammation
3.2. Idiosyncratic DILI: Drug-Specific T Cell Response Triggered by Dendritic Cells
3.3. DILI from Immune-Checkpoint Inhibitors: A Rising Clinical Issue
4. Experimental Methods
4.1. Predictive Models
4.2. In Vitro Models
4.3. In Vivo Models
5. Conclusions

Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AE | adverse event |
| AI-DILI | autoimmune DILI |
| AIH | autoimmune hepatitis |
| ANA | antinuclear antigen |
| APC | antigen-presenting cell |
| APAP | acetaminophen |
| ATP | adenosine triphosphate |
| CD | cluster of differentiation |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| DAMP | damage-associated molecular pattern |
| DC | dendritic cell |
| DILI | drug-induced liver injury |
| FasL | Fas ligand |
| GSH | glutathione |
| GWAS | genome-wide association studies |
| HLA | human leukocyte antigen |
| HMGB1 | high mobility group box-1 |
| ICAM | intercellular adhesion molecule |
| ICI | immune checkpoint inhibitor |
| IFN-γ | interferon gamma |
| iDILI | idiosyncratic drug-induced liver injury |
| IL | interleukin |
| iNKT | invariant NKT cell |
| K18 | keratin 18 |
| KC | Kupffer cell |
| LN | lymph node |
| LPS | lipopolysaccharide |
| MHC | major histocompatibility complex |
| MMP | metalloproteinase |
| NKT | natural killer T cell |
| NLR | NOD-like receptor |
| NO | nitric oxide |
| PAMP | pathogen-associated molecular pattern |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death ligand 1 |
| PRS | polygenic risk score |
| PTPN22 | lymphoid-specific protein tyrosine phosphatase non-receptor type 22 |
| RLR | RIG-like receptor |
| ROS | reactive oxygen species |
| SMA | smooth muscle antigen |
| SNP | single nucleotide polymorphism |
| SOD | superoxide dismutase |
| TCR | T cell receptor |
| Th | T helper cell |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor alpha |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Treg | regulatory T cell |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
References
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef]
- Stephens, C.; Robles-Diaz, M.; Medina-Caliz, I.; Garcia-Cortes, M.; Ortega-Alonso, A.; Sanabria-Cabrera, J.; Gonzalez-Jimenez, A.; Alvarez-Alvarez, I.; Slim, M.; Jimenez-Perez, M.; et al. Comprehensive analysis and insights gained from long-term experience of the Spanish DILI registry. J. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.; Andrade, R.; Aithal, G.; Björnsson, E.; Kaplowitz, N.; Kullak-Ublick, G.; Larrey, D.; Karlsen, T. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An update. Arch. Toxicol. 2020, 94, 3381–3407. [Google Scholar] [CrossRef]
- Björnsson, E.; Talwalkar, J.; Treeprasertsuk, S.; Kamath, P.S.; Takahashi, N.; Sanderson, S.; Neuhauser, M.; Lindor, K. Drug-induced autoimmune hepatitis: Clinical characteristics and prognosis. Hepatology 2010, 51, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Benesic, A.; Rotter, I.; Gerbes, A.L. Early ALT response to corticosteroid treatment distinguishes idiosyncratic drug-induced liver injury from autoimmune hepatitis. Liver Int. 2019, 39, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- de Boer, Y.S.; Kosinski, A.S.; Urban, T.J.; Zhao, Z.; Long, N.; Chalasani, N.; Kleiner, D.E.; Hoofnagle, J.H. Features of Autoimmune Hepatitis in Patients With Drug-induced Liver Injury. Clin. Gastroenterol. Hepatol. 2017, 15, 103–112.e2. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Andrade, R.J. Genetic Predisposition to Drug-Induced Liver Injury. Clin. Liver Dis. 2020, 24, 11–23. [Google Scholar] [CrossRef]
- Nicoletti, P.; Aithal, G.P.; Bjornsson, E.S.; Andrade, R.J.; Sawle, A.; Arrese, M.; Barnhart, H.X.; Bondon-Guitton, E.; Hayashi, P.H.; Bessone, F.; et al. Association of Liver Injury From Specific Drugs, or Groups of Drugs, With Polymorphisms in HLA and Other Genes in a Genome-Wide Association Study. Gastroenterology 2017, 152, 1078–1089. [Google Scholar] [CrossRef]
- Parham, L.R.; Briley, L.P.; Li, L.; Shen, J.; Newcombe, P.J.; King, K.S.; Slater, A.J.; Dilthey, A.; Iqbal, Z.; McVean, G.; et al. Comprehensive genome-wide evaluation of lapatinib-induced liver injury yields a single genetic signal centered on known risk allele HLA-DRB1*07:01. Pharm. J. 2016, 16, 180–185. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Nicoletti, P.; Abramson, K.; Andrade, R.J.; Bjornsson, E.S.; Chalasani, N.; Fontana, R.J.; Hallberg, P.; Li, Y.J.; Lucena, M.I.; et al. A Missense Variant in PTPN22 is a Risk Factor for Drug-induced Liver Injury. Gastroenterology 2019, 156, 1707–1716.e2. [Google Scholar] [CrossRef]
- Stanford, S.M.; Bottini, N. PTPN22: The archetypal non-HLA autoimmunity gene. Nat. Rev. Rheumatol. 2014, 10, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Nielsen, J.; Burn, G.L. A switch-variant model integrates the functions of an autoimmune variant of the phosphatase PTPN22. Sci. Signal. 2018, 11, eaat0936. [Google Scholar] [CrossRef] [PubMed]
- Koido, M.; Kawakami, E.; Fukumura, J.; Noguchi, Y.; Ohori, M.; Nio, Y.; Nicoletti, P.; Aithal, G.P.; Daly, A.K.; Watkins, P.B.; et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nat. Med. 2020, 26, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Kubes, P. Innate Immune Cell Trafficking and Function During Sterile Inflammation of the Liver. Gastroenterology 2016, 151, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Mosedale, M.; Watkins, P.B. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin. Pharmacol. Ther. 2017, 101, 469–480. [Google Scholar] [CrossRef]
- Janeway, C.A.J.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Biomarkers of drug-induced liver injury. Adv. Pharmacol. 2019, 85, 221–239. [Google Scholar] [CrossRef]
- Foureau, D.M.; Walling, T.L.; Maddukuri, V.; Anderson, W.; Culbreath, K.; Kleiner, D.E.; Ahrens, W.A.; Jacobs, C.; Watkins, P.B.; Fontana, R.J.; et al. Comparative analysis of portal hepatic infiltrating leucocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Clin. Exp. Immunol. 2015, 180, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Alhaddad, O.; Elsabaawy, M.; Abdelsameea, E.; Abdallah, A.; Shabaan, A.; Ehsan, N.; Elrefaey, A.; Elsabaawy, D.; Salama, M. Presentations, Causes and Outcomes of Drug-Induced Liver Injury in Egypt. Sci. Rep. 2020, 10, 5124. [Google Scholar] [CrossRef]
- Antoniades, C.G.; Quaglia, A.; Taams, L.S.; Mitry, R.R.; Hussain, M.; Abeles, R.; Possamai, L.A.; Bruce, M.; McPhail, M.; Starling, C.; et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012, 56, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.P.; Cheng, L.; Ju, C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008, 84, 1410–1421. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef]
- Zhang, C.; Feng, J.; Du, J.; Zhuo, Z.; Yang, S.; Zhang, W.; Wang, W.; Zhang, S.; Iwakura, Y.; Meng, G.; et al. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell. Mol. Immunol. 2018, 15, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Pfeilschifter, J.; Mühl, H. A Prominent Role of Interleukin-18 in Acetaminophen-Induced Liver Injury Advocates Its Blockage for Therapy of Hepatic Necroinflammation. Front. Immunol. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, H.A.; Clavien, P.-A. Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology 2002, 122, 202–210. [Google Scholar] [CrossRef]
- Bradham, C.A.; Plümpe, J.; Manns, M.P.; Brenner, D.A.; Trautwein, C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am. J. Physiol. 1998, 275, G387–G392. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Webber, E.M.; Kirillova, I.; Peschon, J.J.; Fausto, N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: Requirement for type 1 but not type 2 receptor. Hepatology 1998, 28, 959–970. [Google Scholar] [CrossRef]
- Selzner, N.; Selzner, M.; Odermatt, B.; Tian, Y.; Van Rooijen, N.; Clavien, P.-A. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology 2003, 124, 692–700. [Google Scholar] [CrossRef]
- Chiu, H.; Gardner, C.R.; Dambach, D.M.; Durham, S.K.; Brittingham, J.A.; Laskin, J.D.; Laskin, D.L. Role of tumor necrosis factor receptor 1 (p55) in hepatocyte proliferation during acetaminophen-induced toxicity in mice. Toxicol. Appl. Pharmacol. 2003, 193, 218–227. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, J.; Jing, Y.; Liu, W.; Yang, X.; Hou, X.; Gao, L.; Wei, L. The concentration of tumor necrosis factor-α determines its protective or damaging effect on liver injury by regulating Yap activity. Cell Death Dis. 2020, 11, 70. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Hammond, K.J.; Poulton, L.D.; Smyth, M.J.; Baxter, A.G. NKT cells: Facts, functions and fallacies. Immunol. Today 2000, 21, 573–583. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef]
- Lee, K.-A.; Kang, M.-H.; Lee, Y.-S.; Kim, Y.-J.; Kim, D.-H.; Ko, H.-J.; Kang, C.-Y. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell. Immunol. 2008, 251, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Kon, S.; Iwabuchi, K.; Kimura, C.; Morimoto, J.; Ito, D.; Segawa, T.; Maeda, M.; Hamuro, J.; Nakayama, T.; et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity 2004, 21, 539–550. [Google Scholar] [CrossRef]
- Laan, M.; Cui, Z.H.; Hoshino, H.; Lötvall, J.; Sjöstrand, M.; Gruenert, D.C.; Skoogh, B.E.; Lindén, A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 1999, 162, 2347–2352. [Google Scholar]
- Wang, X.; Sun, R.; Wei, H.; Tian, Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology 2013, 57, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; You, Q.; Yin, H.; Holt, M.P.; Ju, C. Involvement of natural killer T cells in halothane-induced liver injury in mice. Biochem. Pharmacol. 2010, 80, 255–261. [Google Scholar] [CrossRef]
- Mizrahi, M.; Adar, T.; Lalazar, G.; Nachman, D.; El Haj, M.; Ben Ya’acov, A.; Lichtenstein, Y.; Shabat, Y.; Kanovich, D.; Zolotarov, L.; et al. Glycosphingolipids Prevent APAP and HMG-CoA Reductase Inhibitors-mediated Liver Damage: A Novel Method for “Safer Drug” Formulation that Prevents Drug-induced Liver Injury. J. Clin. Transl. Hepatol. 2018, 6, 127–134. [Google Scholar] [CrossRef]
- Downs, I.; Aw, T.Y.; Liu, J.; Adegboyega, P.; Ajuebor, M.N. Vα14iNKT cell deficiency prevents acetaminophen-induced acute liver failure by enhancing hepatic glutathione and altering APAP metabolism. Biochem. Biophys. Res. Commun. 2012, 428, 245–251. [Google Scholar] [CrossRef][Green Version]
- Martin-Murphy, B.V.; Kominsky, D.J.; Orlicky, D.J.; Donohue, T.M.J.; Ju, C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology 2013, 57, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Smith, C.W. Mechanisms of neutrophil-induced parenchymal cell injury. J. Leukoc. Biol. 1997, 61, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Bajt, M.L. Critical role of CXC chemokines in endotoxemic liver injury in mice. J. Leukoc. Biol. 2004, 76, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, S.K.; Jaeschke, H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef]
- de Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1083–G1088. [Google Scholar] [CrossRef]
- Williams, C.D.; Bajt, M.L.; Sharpe, M.R.; McGill, M.R.; Farhood, A.; Jaeschke, H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol. Appl. Pharmacol. 2014, 275, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Brempelis, K.J.; Crispe, I.N. Infiltrating monocytes in liver injury and repair. Clin. Transl. Immunol. 2016, 5, e113. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.G.; Khamri, W.; Abeles, R.D.; Taams, L.S.; Triantafyllou, E.; Possamai, L.A.; Bernsmeier, C.; Mitry, R.R.; O’Brien, A.; Gilroy, D.; et al. Secretory leukocyte protease inhibitor: A pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology 2014, 59, 1564–1576. [Google Scholar] [CrossRef]
- Wuillemin, N.; Terracciano, L.; Beltraminelli, H.; Schlapbach, C.; Fontana, S.; Krähenbühl, S.; Pichler, W.J.; Yerly, D. T cells infiltrate the liver and kill hepatocytes in HLA-B(∗)57:01-associated floxacillin-induced liver injury. Am. J. Pathol. 2014, 184, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Saide, K.; Farrell, J.; Faulkner, L.; Tailor, A.; Ogese, M.; Daly, A.K.; Pirmohamed, M.; Park, B.K.; Naisbitt, D.J. Characterization of amoxicillin- and clavulanic acid-specific T cells in patients with amoxicillin-clavulanate-induced liver injury. Hepatology 2015, 62, 887–899. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol. Rev. 2000, 177, 134–140. [Google Scholar] [CrossRef]
- Ogese, M.O.; Faulkner, L.; Jenkins, R.E.; French, N.S.; Copple, I.M.; Antoine, D.J.; Elmasry, M.; Malik, H.; Goldring, C.E.; Park, B.K.; et al. Characterization of Drug-Specific Signaling Between Primary Human Hepatocytes and Immune Cells. Toxicol. Sci. 2017, 158, 76–89. [Google Scholar] [CrossRef]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8(+) T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef]
- Rubinstein, D.; Roska, A.K.; Lipsky, P.E. Antigen presentation by liver sinusoidal lining cells after antigen exposure in vivo. J. Immunol. 1987, 138, 1377–1382. [Google Scholar] [PubMed]
- Hanafusa, H.; Morikawa, Y.; Uehara, T.; Kaneto, M.; Ono, A.; Yamada, H.; Ohno, Y.; Urushidani, T. Comparative gene and protein expression analyses of a panel of cytokines in acute and chronic drug-induced liver injury in rats. Toxicology 2014, 324, 43–54. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Knolle, P.A.; Stabenow, D. Mechanisms balancing tolerance and immunity in the liver. Dig. Dis. 2011, 29, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef]
- Padovan, E.; Bauer, T.; Tongio, M.M.; Kalbacher, H.; Weltzien, H.U. Penicilloyl peptides are recognized as T cell antigenic determinants in penicillin allergy. Eur. J. Immunol. 1997, 27, 1303–1307. [Google Scholar] [CrossRef]
- Watkins, S.; Pichler, W.J. Sulfamethoxazole induces a switch mechanism in T cell receptors containing TCRVβ20-1, altering pHLA recognition. PLoS ONE 2013, 8, e76211. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.J.R.; Tuomisto, J.E.E.; Purcell, A.W.; Mifsud, N.A.; Illing, P.T. The complexity of T cell-mediated penicillin hypersensitivity reactions. Allergy 2021, 76, 150–167. [Google Scholar] [CrossRef]
- Padovan, E.; Mauri-Hellweg, D.; Pichler, W.J.; Weltzien, H.U. T cell recognition of penicillin G: Structural features determining antigenic specificity. Eur. J. Immunol. 1996, 26, 42–48. [Google Scholar] [CrossRef]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Tailor, A.; Meng, X.; Adair, K.; Farrell, J.; Waddington, J.C.; Daly, A.; Pirmohamed, M.; Dear, G.; Park, B.K.; Naisbitt, D.J. HLA DRB1*15:01-DQB1*06:02-Restricted Human CD4+ T Cells Are Selectively Activated with Amoxicillin-Peptide Adducts. Toxicol. Sci. 2020, 178, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Monshi, M.M.; Faulkner, L.; Gibson, A.; Jenkins, R.E.; Farrell, J.; Earnshaw, C.J.; Alfirevic, A.; Cederbrant, K.; Daly, A.K.; French, N.; et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology 2013, 57, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.; Hammond, S.; Jaruthamsophon, K.; Roth, S.; Mosedale, M.; Naisbitt, D.J. Tolvaptan- and Tolvaptan-Metabolite-Responsive T Cells in Patients with Drug-Induced Liver Injury. Chem. Res. Toxicol. 2020, 33, 2745–2748. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Xiang, X.; Mo, R.; Bao, R.; Wang, P.; Guo, S.; Zhao, G.; Gui, H.; Wang, H.; Bao, S.; et al. Protective effect of Th22 cells and intrahepatic IL-22 in drug induced hepatocellular injury. J. Hepatol. 2015, 63, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Cottagiri, M.; Nyandjo, M.; Stephens, M.; Mantilla, J.J.; Saito, H.; Mackay, I.R.; Rose, N.R.; Njoku, D.B. In drug-induced, immune-mediated hepatitis, interleukin-33 reduces hepatitis and improves survival independently and as a consequence of FoxP3+ T-cell activity. Cell. Mol. Immunol. 2019, 16, 706–717. [Google Scholar] [CrossRef]
- deLemos, A.S.; Foureau, D.M.; Jacobs, C.; Ahrens, W.; Russo, M.W.; Bonkovsky, H.L. Drug-induced liver injury with autoimmune features. Semin. Liver Dis. 2014, 34, 194–204. [Google Scholar] [CrossRef]
- Metushi, I.; Uetrecht, J.; Phillips, E. Mechanism of isoniazid-induced hepatotoxicity: Then and now. Br. J. Clin. Pharmacol. 2016, 81, 1030–1036. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef]
- Di Rosa, F.; Serafini, B.; Scognamiglio, P.; Di Virgilio, A.; Finocchi, L.; Aloisi, F.; Barnaba, V. Short-lived immunization site inflammation in self-limited active experimental allergic encephalomyelitis. Int. Immunol. 2000, 12, 711–719. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Badros, A.; Hyjek, E.; Ma, N.; Lesokhin, A.; Dogan, A.; Rapoport, A.P.; Kocoglu, M.; Lederer, E.; Philip, S.; Milliron, T.; et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 130, 1189–1197. [Google Scholar] [CrossRef]
- Nayak, L.; Iwamoto, F.M.; LaCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet. Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Agostinetto, E.; Eiger, D.; Lambertini, M.; Ceppi, M.; Bruzzone, M.; Pondé, N.; Plummer, C.; Awada, A.H.; Santoro, A.; Piccart-Gebhart, M.; et al. Cardiotoxicity of immune checkpoint inhibitors: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 148, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Wolchok, J.D.; Yosipovitch, G.; Kähler, K.C.; Busam, K.J.; Hauschild, A. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J. Am. Acad. Dermatol. 2014, 71, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Eigentler, T.K.; Hassel, J.C.; Berking, C.; Aberle, J.; Bachmann, O.; Grünwald, V.; Kähler, K.C.; Loquai, C.; Reinmuth, N.; Steins, M.; et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat. Rev. 2016, 45, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Barnabei, A.; Marchetti, P.; De Vecchis, L.; Salvatori, R.; Torino, F. Endocrine side effects induced by immune checkpoint inhibitors. J. Clin. Endocrinol. Metab. 2013, 98, 1361–1375. [Google Scholar] [CrossRef]
- Kim, K.W.; Ramaiya, N.H.; Krajewski, K.M.; Jagannathan, J.P.; Tirumani, S.H.; Srivastava, A.; Ibrahim, N. Ipilimumab associated hepatitis: Imaging and clinicopathologic findings. Investig. New Drugs 2013, 31, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Postow, M.; Lao, C.D.; Schadendorf, D. Management of Adverse Events Following Treatment With Anti-Programmed Death-1 Agents. Oncologist 2016, 21, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Iorgulescu, J.B.; Braun, D.; Oliveira, G.; Keskin, D.B.; Wu, C.J. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 2018, 10, 87. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- De Martin, E.; Michot, J.-M.; Rosmorduc, O.; Guettier, C.; Samuel, D. Liver toxicity as a limiting factor to the increasing use of immune checkpoint inhibitors. JHEP Rep. 2020, 2, 100170. [Google Scholar] [CrossRef]
- Gudd, C.L.C.; Au, L.; Triantafyllou, E.; Shum, B.; Liu, T.; Nathwani, R.; Kumar, N.; Mukherjee, S.; Dhar, A.; Woollard, K.J.; et al. Activation and transcriptional profile of monocytes and CD8+ T cells are altered in checkpoint inhibitor-related hepatitis. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- Zen, Y.; Yeh, M.M. Hepatotoxicity of immune checkpoint inhibitors: A histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod. Pathol. 2018, 31, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Halliday, N.; Carbone, M.; Invernizzi, P.; Thorburn, D. Open challenges in the management of autoimmune hepatitis. Minerva Gastroenterol. Dietol. 2020. [Google Scholar] [CrossRef]
- Kole, C.; Charalampakis, N.; Tsakatikas, S.; Vailas, M.; Moris, D.; Gkotsis, E.; Kykalos, S.; Karamouzis, M.V.; Schizas, D. Immunotherapy for Hepatocellular Carcinoma: A 2021 Update. Cancers 2020, 12, 2859. [Google Scholar] [CrossRef]
- Babai, S.; Auclert, L.; Le-Louët, H. Safety data and withdrawal of hepatotoxic drugs. Therapie 2018. [Google Scholar] [CrossRef] [PubMed]
- Katarey, D.; Verma, S. Drug-induced liver injury. Clin. Med. 2016, 16, s104–s109. [Google Scholar] [CrossRef]
- Chen, M.; Bisgin, H.; Tong, L.; Hong, H.; Fang, H.; Borlak, J.; Tong, W. Toward predictive models for drug-induced liver injury in humans: Are we there yet? Biomark. Med. 2014, 8, 201–213. [Google Scholar] [CrossRef]
- Iorga, A.; Dara, L. Cell Death in Drug-Induced Liver Injury, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 85, ISBN 9780128167595. [Google Scholar]
- Lammert, C.; Einarsson, S.; Saha, C.; Niklasson, A.; Bjornsson, E.; Chalasani, N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 2008, 47, 2003–2009. [Google Scholar] [CrossRef]
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the challenge of drug-induced liver injury: A roadmap for the development and deployment of preclinical predictive models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of Drug Induced Liver Injury (DILI)—Current Issues and Future Perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef]
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S.; Cagide Fajin, J.L.; Morell, C.; Ruiz, R.M.; Cañizares-Carmenate, Y.; Dominguez, E.R. Desirability-based methods of multiobjective optimization and ranking for global QSAR studies. Filtering safe and potent drug candidates from combinatorial libraries. J. Comb. Chem. 2008, 10, 897–913. [Google Scholar] [CrossRef]
- Tolosa, L.; Pinto, S.; Donato, M.T.; Lahoz, A.; Castell, J.V.; O’connor, J.E.; Gómez-Lechón, M.J. Development of a multiparametric cell-based protocol to screen and classify the hepatotoxicity potential of drugs. Toxicol. Sci. 2012, 127, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Diaz, D.; O’Brien, P.J. Applications of cytotoxicity assays and pre-lethal mechanistic assays for assessment of human hepatotoxicity potential. Chem. Biol. Interact. 2004, 150, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.; Bruckner, D.M.; Dordick, J.S. Advancing Predictive Hepatotoxicity at the Intersection of Experimental, in Silico, and Artificial Intelligence Technologies. Chem. Res. Toxicol. 2018, 31, 412–430. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip’. Exp. Cell Res. 2018, 363, 15–25. [Google Scholar] [CrossRef]
- Tomida, T.; Okamura, H.; Satsukawa, M.; Yokoi, T.; Konno, Y. Multiparametric assay using HepaRG cells for predicting drug-induced liver injury. Toxicol. Lett. 2015, 236, 16–24. [Google Scholar] [CrossRef]
- Wu, Y.; Geng, X.C.; Wang, J.F.; Miao, Y.F.; Lu, Y.; Li, B. The HepaRG cell line, a superior in vitro model to L-02, HepG2 and hiHeps cell lines for assessing drug-induced liver injury. Cell Biol. Toxicol. 2016, 32, 37–59. [Google Scholar] [CrossRef]
- Le Vee, M.; Noel, G.; Jouan, E.; Stieger, B.; Fardel, O. Polarized expression of drug transporters in differentiated human hepatoma HepaRG cells. Toxicol. Vitr. 2013, 27, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Yi, F.; Liu, G.H.; Belmonte, J.C.I. Human induced pluripotent stem cells derived hepatocytes: Rising promise for disease modeling, drug development and cell therapy. Protein Cell 2012, 3, 246–250. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Fleming, H.E.; Khetani, S.R.; Bhatia, S.N. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol. Adv. 2014, 32, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Tolosa, L. Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 2019, 106, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ware, B.R.; Berger, D.R.; Khetani, S.R. Prediction of drug-induced liver injury in micropatterned co-cultures containing iPSC-derived human hepatocytes. Toxicol. Sci. 2015, 145, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Balis, U.J.; Yarmush, M.L.; Toner, M. Effect of cell–cell interactions in preservation of cellular phenotype: Cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 1999, 13, 1883–1900. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, Y.S.; Culberson, C.R.; Coger, R.N. Contribution of non-parenchymal cells to the performance of micropatterned hepatocytes. Tissue Eng. 2006, 12, 2241–2251. [Google Scholar] [CrossRef]
- Khetani, S.R.; Kanchagar, C.; Ukairo, O.; Krzyzewski, S.; Moore, A.; Shi, J.; Aoyama, S.; Aleo, M.; Will, Y. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol. Sci. 2013, 132, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Smith, L.; Bharadwaj, S.; Atala, A.; Soker, S.; Zhang, Y. Tissue specific synthetic ECM hydrogels for 3-D in vitro maintenance of hepatocyte function. Biomaterials 2012, 33, 4565–4575. [Google Scholar] [CrossRef] [PubMed]
- Novik, E.; Maguire, T.J.; Chao, P.; Cheng, K.C.; Yarmush, M.L. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochem. Pharmacol. 2010, 79, 1036–1044. [Google Scholar] [CrossRef]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, functional, and mechanistic comparisons of stem cell-derived hepatocytes, HepaRG cells, and three-dimensional human hepatocyte spheroids as predictive in vitro systems for drug-induced liver injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Proctor, W.R.; Foster, A.J.; Vogt, J.; Summers, C.; Middleton, B.; Pilling, M.A.; Shienson, D.; Kijanska, M.; Ströbel, S.; Kelm, J.M.; et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol. 2017, 91, 2849–2863. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of hepatic 2D sandwich cultures and 3d spheroids for long-term toxicity applications: A multicenter study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Ullah, S.; Schmidt, S.; Nandania, J.; Velagapudi, V.; Beck, O.; Ingelman-Sundberg, M.; Lauschke, V.M. Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J. 2017, 31, 2696–2708. [Google Scholar] [CrossRef]
- Vinci, B.; Duret, C.; Klieber, S.; Gerbal-Chaloin, S.; Sa-Cunha, A.; Laporte, S.; Suc, B.; Maurel, P.; Ahluwalia, A.; Daujat-Chavanieu, M. Modular bioreactor for primary human hepatocyte culture: Medium flow stimulates expression and activity of detoxification genes. Biotechnol. J. 2011, 6, 554–564. [Google Scholar] [CrossRef]
- Hoffmann, S.A.; Müller-Vieira, U.; Biemel, K.; Knobeloch, D.; Heydel, S.; Lübberstedt, M.; Nüssler, A.K.; Andersson, T.B.; Gerlach, J.C.; Zeilinger, K. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol. Bioeng. 2012, 109, 3172–3181. [Google Scholar] [CrossRef]
- Tostões, R.M.; Leite, S.B.; Serra, M.; Jensen, J.; Björquist, P.; Carrondo, M.J.T.; Brito, C.; Alves, P.M. Human liver cell spheroids in extended perfusion bioreactor culture for repeated-dose drug testing. Hepatology 2012, 55, 1227–1236. [Google Scholar] [CrossRef]
- Vernetti, L.A.; Senutovitch, N.; Boltz, R.; DeBiasio, R.; Ying Shun, T.; Gough, A.; Taylor, D.L. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp. Biol. Med. 2016, 241, 101–114. [Google Scholar] [CrossRef]
- Gough, A.; Soto-Gutierrez, A.; Vernetti, L.; Ebrahimkhani, M.R.; Stern, A.M.; Taylor, D.L. Human biomimetic liver microphysiology systems in drug development and precision medicine. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Morita, M.; Hosomi, H.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Knockdown of superoxide dismutase 2 enhances acetaminophen-induced hepatotoxicity in rat. Toxicology 2009, 264, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Davis, J.S.; Woolbright, B.L.; Du, K.; Cahkraborty, M.; Weemhoff, J.; Jaeschke, H.; Bourdi, M. Differential susceptibility to acetaminophen-induced liver injury in sub-strains of C57BL/6 mice: 6N versus 6J. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2016, 98, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Yohe, H.C.; O’Hara, K.A.; Hunt, J.A.; Kitzmiller, T.J.; Wood, S.G.; Bement, J.L.; Bement, W.J.; Szakacs, J.G.; Wrighton, S.A.; Jacobs, J.M.; et al. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am. J. Physiol. Liver Physiol. 2006, 290, G1269–G1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-X.; Han, D.; Gunawan, B.; Kaplowitz, N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006, 43, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kim, J.-W.; Zhou, Z.; Qi, J.; Tian, W.; Lim, C.W.; Han, K.M.; Kim, B. Macrophage-Inducible C-Type Lectin Signaling Exacerbates Acetaminophen-Induced Liver Injury by Promoting Kupffer Cell Activation in Mice. Mol. Pharmacol. 2021, 99, 92–103. [Google Scholar] [CrossRef]
- Buchweitz, J.P.; Ganey, P.E.; Bursian, S.J.; Roth, R.A. Underlying endotoxemia augments toxic responses to chlorpromazine: Is there a relationship to drug idiosyncrasy? J. Pharmacol. Exp. Ther. 2002, 300, 460–467. [Google Scholar] [CrossRef]
- Lu, J.; Jones, A.D.; Harkema, J.R.; Roth, R.A.; Ganey, P.E. Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: Role of tumor necrosis factor-alpha. Toxicol. Sci. 2012, 125, 126–133. [Google Scholar] [CrossRef]
- Shaw, P.J.; Hopfensperger, M.J.; Ganey, P.E.; Roth, R.A. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol. Sci. 2007, 100, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Metushi, I.G.; Hayes, M.A.; Uetrecht, J. Treatment of PD-1(-/-) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015, 61, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Uetrecht, J. The Combination of Anti-CTLA-4 and PD1-/- Mice Unmasks the Potential of Isoniazid and Nevirapine To Cause Liver Injury. Chem. Res. Toxicol. 2015, 28, 2287–2291. [Google Scholar] [CrossRef]
- Cho, T.; Wang, X.; Yeung, K.; Cao, Y.; Uetrecht, J. Liver Injury Caused by Green Tea Extract in PD-1(-/-) Mice: An Impaired Immune Tolerance Model for Idiosyncratic Drug-Induced Liver Injury. Chem. Res. Toxicol. 2021, 34, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Uetrecht, J. The Role of CD8 T Cells in Amodiaquine-Induced Liver Injury in PD1-/- Mice Cotreated with Anti-CTLA-4. Chem. Res. Toxicol. 2015, 28, 1567–1573. [Google Scholar] [CrossRef]
- Ong, M.M.K.; Latchoumycandane, C.; Boelsterli, U.A. Troglitazone-induced hepatic necrosis in an animal model of silent genetic mitochondrial abnormalities. Toxicol. Sci. 2007, 97, 205–213. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerussi, A.; Natalini, A.; Antonangeli, F.; Mancuso, C.; Agostinetto, E.; Barisani, D.; Di Rosa, F.; Andrade, R.; Invernizzi, P. Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. Int. J. Mol. Sci. 2021, 22, 4557. https://doi.org/10.3390/ijms22094557
Gerussi A, Natalini A, Antonangeli F, Mancuso C, Agostinetto E, Barisani D, Di Rosa F, Andrade R, Invernizzi P. Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. International Journal of Molecular Sciences. 2021; 22(9):4557. https://doi.org/10.3390/ijms22094557
Chicago/Turabian StyleGerussi, Alessio, Ambra Natalini, Fabrizio Antonangeli, Clara Mancuso, Elisa Agostinetto, Donatella Barisani, Francesca Di Rosa, Raul Andrade, and Pietro Invernizzi. 2021. "Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models" International Journal of Molecular Sciences 22, no. 9: 4557. https://doi.org/10.3390/ijms22094557
APA StyleGerussi, A., Natalini, A., Antonangeli, F., Mancuso, C., Agostinetto, E., Barisani, D., Di Rosa, F., Andrade, R., & Invernizzi, P. (2021). Immune-Mediated Drug-Induced Liver Injury: Immunogenetics and Experimental Models. International Journal of Molecular Sciences, 22(9), 4557. https://doi.org/10.3390/ijms22094557