
Lymphopenia, Lymphopenia-Induced Proliferation, and Autoimmunity


Abstract
1. Introduction
2. Immune Homeostasis
3. Lymphopenia in Physiological and Autoimmune Conditions
3.1. Lymphopenia in Human Autoimmune Diseases
3.2. Lymphopenia in Autoimmune-Prone Animal Models
4. LIP Is Associated with Autoimmunity in Both Animal Models and Humans
4.1. LIP-Associated Autoimmunity in Animal Models
4.2. LIP-Associated Autoimmunity in Humans
4.3. Setting in Which LIP Causes Autoimmune Diseases
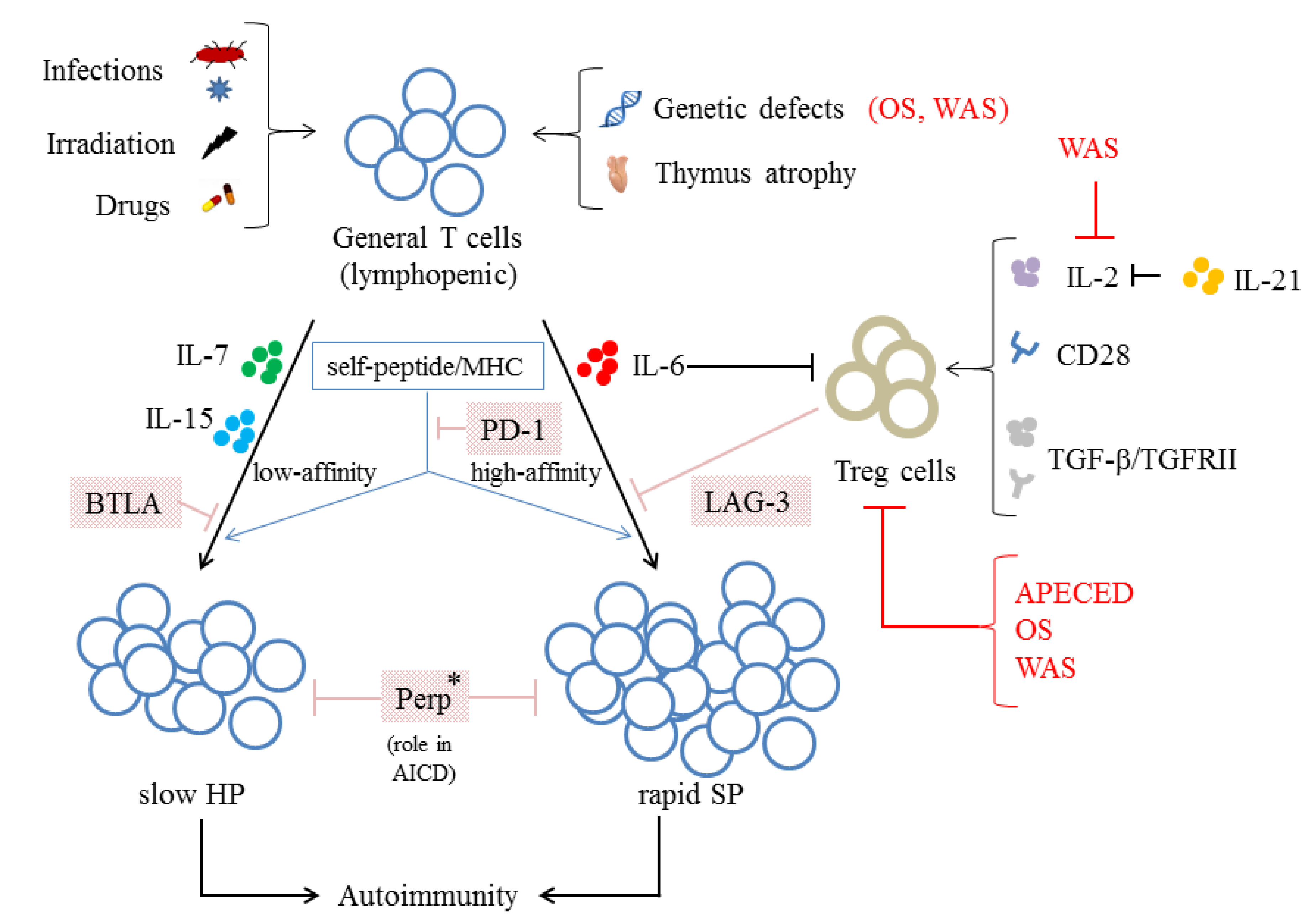
5. Lymphopenia Alone Is Not Sufficient to Induce Autoimmunity
6. The Loss of Control over Homeostasis
6.1. The Relationship between the Loss of Control over Homeostasis and the Function/Number of Treg Cells
6.2. Other Mechanisms Negatively Regulate the LIP of T Cells
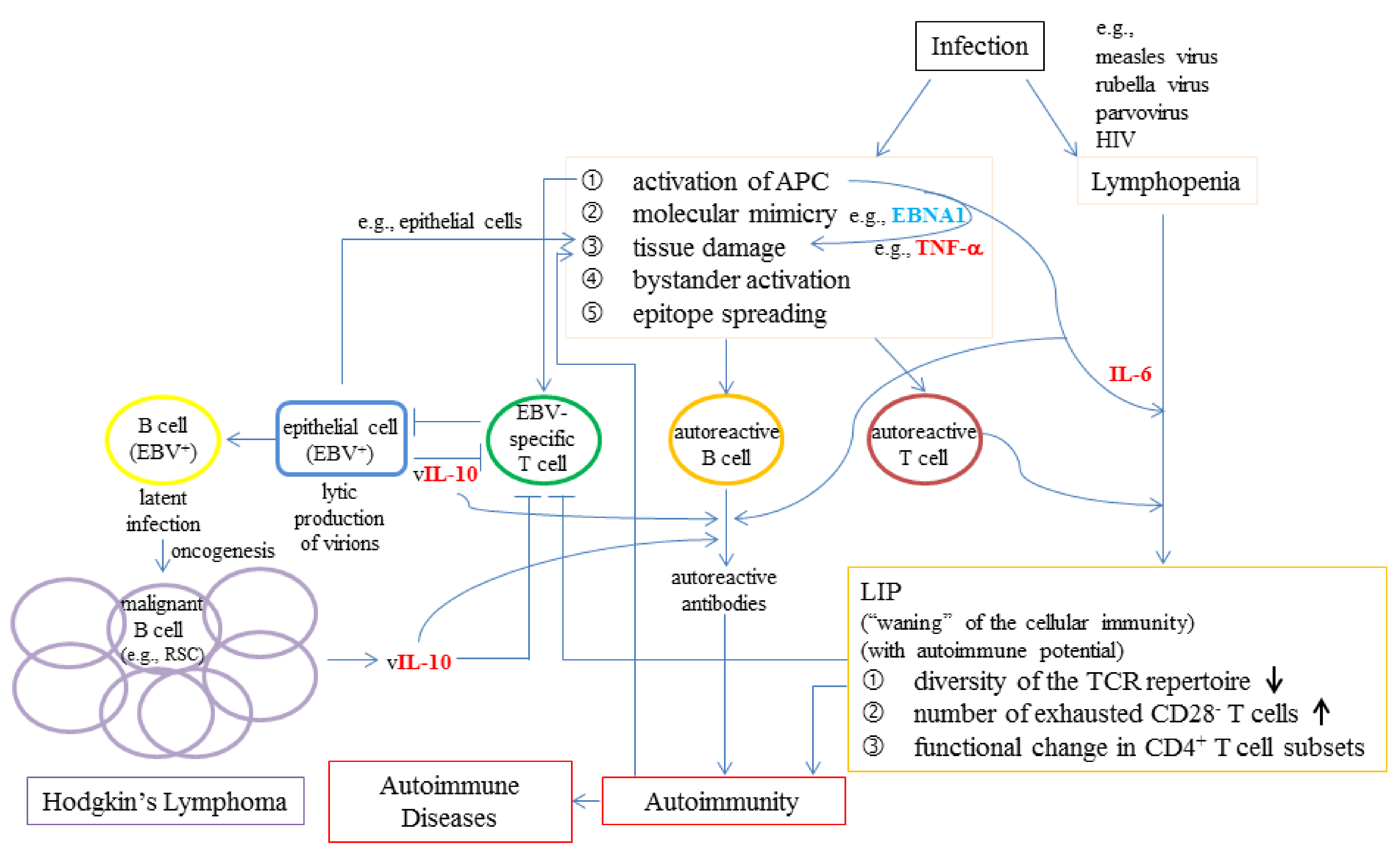
6.3. The Possible Roles of Viral Infections in LIP, Autoimmunity, and Lymphoproliferative Conditions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| ADs | autoimmune diseases |
| ADAP | adhesion and degranulation-promoting adapter protein |
| AIRE | autoimmune regulator |
| APCs | antigen-presenting cells |
| APECED | autoimmune polyendocrinopathy candidiasis ectodermal dystrophy |
| Bcl-6 | B-cell lymphoma 6 |
| βgal | β-galactosidase |
| BL | Burkitt’s lymphoma |
| Breg | regulatory B cells |
| BTLA | B and T lymphocyte attenuator |
| CFA | complete Freund’s adjuvant |
| CMV | cytomegalovirus |
| COVID-19 | coronavirus disease 2019 |
| CTLs | cytotoxic T lymphocytes |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CTX | cyclophosphamide |
| CVID | common variable immunodeficiency |
| CXCR5 | C-X-C chemokine receptor type 5 |
| DCs | dendritic cells |
| DCLRE1C | deoxyribonucleic acid cross-link repair 1C |
| DMARD | disease-modifying anti-rheumatic drug |
| EAU | experimental autoimmune uveoretinitis |
| EBNA1 | Epstein–Barr nuclear antigen 1 |
| EBV | Epstein–Barr virus |
| F759 | mice with a mutation in the gp130 IL-6 receptor subunit |
| FK506 | tacrolimus |
| FoxP3 | forkhead box protein transcription factor P3 |
| GAD | glutamate acid decarboxylase |
| HAART | highly active antiretroviral therapy |
| HIV | human immunodeficiency virus |
| HLA | human leukocyte antigen |
| HP | homeostatic proliferation |
| HT | Hashimoto’s thyroiditis |
| HTLV-1 | Human T-lymphotropic virus 1 |
| IFN-γ | interferon-gamma |
| IL | Interleukin |
| IL-2R | IL-2 receptor |
| IL-21R | IL-21 receptor |
| INS | insulin |
| IRIS | immune reconstitution inflammatory syndrome |
| iTreg/pTreg | peripherally-induced Treg cells |
| K/BxN | a cross between KRN TCR transgenic mice on a C57BL/6 background (K/B) and NOD mice |
| LAG-3 | lymphocyte activation gene 3 |
| LCMV | lymphocytic choriomeningitis virus |
| LIG4 | ligase 4 |
| LIP | lymphopenia-induced proliferation |
| LNs | lymph nodes |
| MHC | major histocompatibility complex class |
| MM | memory mutant |
| MTX | methotrexate |
| NK | natural killer cells |
| NKT | natural killer T cells |
| NOD | non-obese diabetic |
| nTreg/tTreg | naturally occurring thymus-derived Treg cells |
| OS | Omenn syndrome |
| PD-1 | programmed cell death protein 1 |
| Perp | p53 apoptosis effector related to PMP-22 |
| PID | primary immunodeficiency |
| PIDs | primary immunodeficiencies |
| PRRs | pattern-recognition receptors |
| pSS | primary Sjögren’s syndrome |
| PTLD | post-transplant lymphoproliferative disorder |
| PTPN2 | protein tyrosine phosphatase N2 |
| RA | rheumatoid arthritis |
| RAG1 | recombination activating gene 1 |
| RAG2 | recombination activating gene 2 |
| RIP-GP | rat insulin promoter-glycoprotein |
| RSCs | Reed-Sternberg cells |
| RTL | relative telomere length |
| RTEs | recent thymic emigrants |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SCID | severe combined immune deficiency |
| SLE | systemic lupus erythematosus |
| SNP | single nucleotide polymorphism |
| SP | spontaneous proliferation |
| SS | Sjögren’s syndrome |
| STAT3 | signal transducer and activator of transcription 3 |
| T1D | type 1 diabetes |
| TCR | T cell receptor |
| Tfh | T follicular helper |
| Tfr | follicular regulatory T cells |
| TGF-β | transforming growth factor-beta |
| TGF-βRII | transforming growth factor-beta receptor 2 |
| tgfbr2 | transforming growth factor-beta receptor 2 |
| Th1 | type 1 helper cells |
| Th3 | type 3 helper cells |
| Th17 | type 17 helper cells |
| TLRs | Toll-like receptors |
| TNF-α | tumor necrosis factor-alpha |
| Tr1 | type 1 regulatory T cells |
| TRECs | T-cell receptor excision circles |
| Tregs | regulatory T cells |
| WAS | Wiskott–Aldrich syndrome |
| WASP | WAS protein |
References
- Akbar, A.N.; Vukmanovic-Stejic, M.; Taams, L.S.; Macallan, D.C. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat. Rev. Immunol. 2007, 7, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Surh, C.D.; Sprent, J. TGF-beta puts the brakes on homeostatic proliferation. Nat. Immunol. 2012, 13, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Lee, J.; Zhou, Y.J.; Schreiber, J.J.; Keler, T.; Puhr, S.; Anandasabapathy, N.; Schlesinger, S.; Caskey, M.; Liu, K.; et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J. Exp. Med. 2015, 212, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Lee, D.S.; Chang, J.M.; Sprent, J.; Surh, C.D. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 1999, 11, 173–181. [Google Scholar] [CrossRef]
- Goldrath, A.W.; Bevan, M.J. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 1999, 11, 183–190. [Google Scholar] [CrossRef]
- Takada, K.; Jameson, S.C. Naive T cell homeostasis: From awareness of space to a sense of place. Nat. Rev. Immunol. 2009, 9, 823–832. [Google Scholar] [CrossRef]
- King, C.; Ilic, A.; Koelsch, K.; Sarvetnick, N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 2004, 117, 265–277. [Google Scholar] [CrossRef]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef]
- Tan, J.T.; Dudl, E.; LeRoy, E.; Murray, R.; Sprent, J.; Weinberg, K.I.; Surh, C.D. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. USA 2001, 98, 8732–8737. [Google Scholar] [CrossRef]
- Martin, B.; Becourt, C.; Bienvenu, B.; Lucas, B. Self-recognition is crucial for maintaining the peripheral CD4+ T-cell pool in a nonlymphopenic environment. Blood 2006, 108, 270–277. [Google Scholar] [CrossRef]
- Kieper, W.C.; Troy, A.; Burghardt, J.T.; Ramsey, C.; Lee, J.Y.; Jiang, H.Q.; Dummer, W.; Shen, H.; Cebra, J.J.; Surh, C.D. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J. Immunol. 2005, 174, 3158–3163. [Google Scholar] [CrossRef]
- Moses, C.T.; Thorstenson, K.M.; Jameson, S.C.; Khoruts, A. Competition for self ligands restrains homeostatic proliferation of naive CD4 T cells. Proc. Natl. Acad. Sci. USA 2003, 100, 1185–1190. [Google Scholar] [CrossRef]
- Tajima, M.; Wakita, D.; Noguchi, D.; Chamoto, K.; Yue, Z.; Fugo, K.; Ishigame, H.; Iwakura, Y.; Kitamura, H.; Nishimura, T. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J. Exp. Med. 2008, 205, 1019–1027. [Google Scholar] [CrossRef]
- Min, B.; Foucras, G.; Meier-Schellersheim, M.; Paul, W.E. Spontaneous proliferation, a response of naive CD4 T cells determined by the diversity of the memory cell repertoire. Proc. Natl. Acad. Sci. USA 2004, 101, 3874–3879. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Yamane, H.; Hu-Li, J.; Paul, W.E. Spontaneous and homeostatic proliferation of CD4 T cells are regulated by different mechanisms. J. Immunol. 2005, 174, 6039–6044. [Google Scholar] [CrossRef]
- Rocha, B.; Freitas, A.A.; Coutinho, A.A. Population dynamics of T lymphocytes. Renewal rate and expansion in the peripheral lymphoid organs. J. Immunol. 1983, 131, 2158–2164. [Google Scholar]
- Tanchot, C.; Rosado, M.M.; Agenes, F.; Freitas, A.A.; Rocha, B. Lymphocyte homeostasis. Semin. Immunol. 1997, 9, 331–337. [Google Scholar] [CrossRef]
- Sprent, J.; Surh, C.D. Cytokines and T cell homeostasis. Immunol. Lett. 2003, 85, 145–149. [Google Scholar] [CrossRef]
- Jameson, S.C. T cell homeostasis: Keeping useful T cells alive and live T cells useful. Semin. Immunol. 2005, 17, 231–237. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Wolkers, M.C.; Schoenberger, S.P.; Jameson, S.C. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat. Immunol. 2006, 7, 475–481. [Google Scholar] [CrossRef]
- Goldrath, A.W.; Bogatzki, L.Y.; Bevan, M.J. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J. Exp. Med. 2000, 192, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.K.; Rao, V.P.; Ge, Q.; Eisen, H.N.; Chen, J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J. Exp. Med. 2000, 192, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Winstead, C.J.; Fraser, J.M.; Khoruts, A. Regulatory CD4+CD25+Foxp3+ T cells selectively inhibit the spontaneous form of lymphopenia-induced proliferation of naive T cells. J. Immunol. 2008, 180, 7305–7317. [Google Scholar] [CrossRef] [PubMed]
- Le Campion, A.; Bourgeois, C.; Lambolez, F.; Martin, B.; Leaument, S.; Dautigny, N.; Tanchot, C.; Penit, C.; Lucas, B. Naive T cells proliferate strongly in neonatal mice in response to self-peptide/self-MHC complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 4538–4543. [Google Scholar] [CrossRef]
- Owen, D.L.; Mahmud, S.A.; Sjaastad, L.E.; Williams, J.B.; Spanier, J.A.; Simeonov, D.R.; Ruscher, R.; Huang, W.; Proekt, I.; Miller, C.N.; et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat. Immunol. 2019, 20, 195–205. [Google Scholar] [CrossRef]
- Bilate, A.M.; Lafaille, J.J. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 2012, 30, 733–758. [Google Scholar] [CrossRef]
- Walker, M.R.; Kasprowicz, D.J.; Gersuk, V.H.; Benard, A.; van Landeghen, M.; Buckner, J.H.; Ziegler, S.F. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J. Clin. Investig. 2003, 112, 1437–1443. [Google Scholar] [CrossRef]
- Cozzo, C.; Larkin, J., III; Caton, A.J. Cutting edge: Self-peptides drive the peripheral expansion of CD4+CD25+ regulatory T cells. J. Immunol. 2003, 171, 5678–5682. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Abbas, A.K. Natural versus adaptive regulatory T cells. Nat. Rev. Immunol. 2003, 3, 253–257. [Google Scholar] [CrossRef]
- Sawant, D.V.; Hamilton, K.; Vignali, D.A. Interleukin-35: Expanding Its Job Profile. J. Interferon Cytokine Res. 2015, 35, 499–512. [Google Scholar] [CrossRef]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef]
- Inobe, J.; Slavin, A.J.; Komagata, Y.; Chen, Y.; Liu, L.; Weiner, H.L. IL-4 is a differentiation factor for transforming growth factor-beta secreting Th3 cells and oral administration of IL-4 enhances oral tolerance in experimental allergic encephalomyelitis. Eur. J. Immunol. 1998, 28, 2780–2790. [Google Scholar] [CrossRef]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef]
- Maceiras, A.R.; Fonseca, V.R.; Agua-Doce, A.; Graca, L. T follicular regulatory cells in mice and men. Immunology 2017, 152, 25–35. [Google Scholar] [CrossRef]
- Lim, H.W.; Hillsamer, P.; Kim, C.H. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J. Clin. Investig. 2004, 114, 1640–1649. [Google Scholar] [CrossRef]
- Haynes, L.M.; Vanderlugt, C.L.; dal Canto, M.C.; Melvold, R.W.; Miller, S.D. CD8(+) T cells from Theiler’s virus-resistant BALB/cByJ mice downregulate pathogenic virus-specific CD4(+) T cells. J. Neuroimmunol. 2000, 106, 43–52. [Google Scholar] [CrossRef]
- Elizondo, D.M.; Andargie, T.E.; Haddock, N.L.; da Silva, R.L.L.; de Moura, T.R.; Lipscomb, M.W. IL-10 producing CD8(+) CD122(+) PD-1(+) regulatory T cells are expanded by dendritic cells silenced for Allograft Inflammatory Factor-1. J. Leukoc. Biol. 2019, 105, 123–130. [Google Scholar] [CrossRef]
- Harrison, L.C.; Dempsey-Collier, M.; Kramer, D.R.; Takahashi, K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J. Exp. Med. 1996, 184, 2167–2174. [Google Scholar] [CrossRef]
- Seo, N.; Tokura, Y.; Takigawa, M.; Egawa, K. Depletion of IL-10- and TGF-beta-producing regulatory gamma delta T cells by administering a daunomycin-conjugated specific monoclonal antibody in early tumor lesions augments the activity of CTLs and NK cells. J. Immunol. 1999, 163, 242–249. [Google Scholar]
- Sonoda, K.H.; Faunce, D.E.; Taniguchi, M.; Exley, M.; Balk, S.; Stein-Streilein, J. NK T cell-derived IL-10 is essential for the differentiation of antigen-specific T regulatory cells in systemic tolerance. J. Immunol. 2001, 166, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.M.; Fadel, S.A.; Cao, S.; Sarzotti, M. T cell immunity in neonates. Immunol. Res. 2000, 22, 177–190. [Google Scholar] [CrossRef]
- Min, B.; McHugh, R.; Sempowski, G.D.; Mackall, C.; Foucras, G.; Paul, W.E. Neonates support lymphopenia-induced proliferation. Immunity 2003, 18, 131–140. [Google Scholar] [CrossRef]
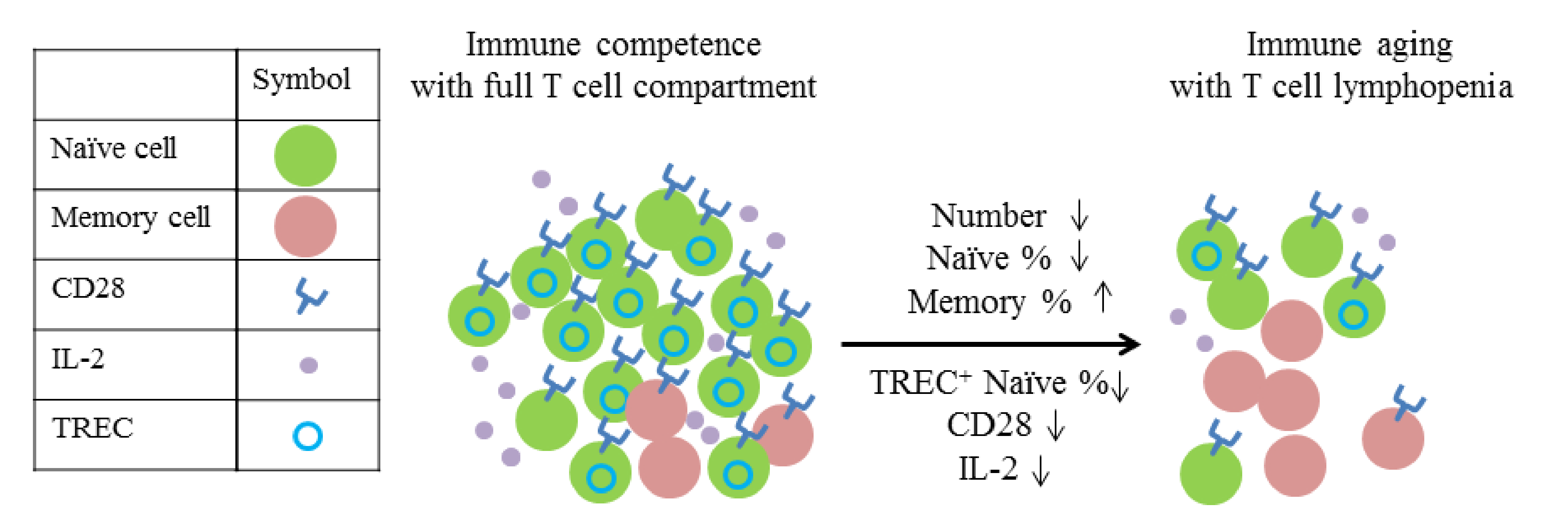
- Naylor, K.; Li, G.; Vallejo, A.N.; Lee, W.W.; Koetz, K.; Bryl, E.; Witkowski, J.; Fulbright, J.; Weyand, C.M.; Goronzy, J.J. The influence of age on T cell generation and TCR diversity. J. Immunol. 2005, 174, 7446–7452. [Google Scholar] [CrossRef]
- Woods, J.A.; Ceddia, M.A.; Zack, M.D.; Lowder, T.W.; Lu, Q. Exercise training increases the naive to memory T cell ratio in old mice. Brain Behav. Immun. 2003, 17, 384–392. [Google Scholar] [CrossRef]
- Sheu, T.T.; Chiang, B.L.; Yen, J.H.; Lin, W.C. Premature CD4+ T cell aging and its contribution to lymphopenia-induced proliferation of memory cells in autoimmune-prone non-obese diabetic mice. PLoS ONE 2014, 9, e89379. [Google Scholar] [CrossRef]
- Prelog, M. Aging of the immune system: A risk factor for autoimmunity? Autoimmun. Rev. 2006, 5, 136–139. [Google Scholar] [CrossRef]
- Koetz, K.; Bryl, E.; Spickschen, K.; O’Fallon, W.M.; Goronzy, J.J.; Weyand, C.M. T cell homeostasis in patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 9203–9208. [Google Scholar] [CrossRef]
- Douek, D.C.; McFarland, R.D.; Keiser, P.H.; Gage, E.A.; Massey, J.M.; Haynes, B.F.; Polis, M.A.; Haase, A.T.; Feinberg, M.B.; Sullivan, J.L.; et al. Changes in thymic function with age and during the treatment of HIV infection. Nature 1998, 396, 690–695. [Google Scholar] [CrossRef]
- Chamberlain, W.D.; Falta, M.T.; Kotzin, B.L. Functional subsets within clonally expanded CD8(+) memory T cells in elderly humans. Clin. Immunol. 2000, 94, 160–172. [Google Scholar] [CrossRef]
- Franceschi, C.; Valensin, S.; Fagnoni, F.; Barbi, C.; Bonafe, M. Biomarkers of immunosenescence within an evolutionary perspective: The challenge of heterogeneity and the role of antigenic load. Exp. Gerontol. 1999, 34, 911–921. [Google Scholar] [CrossRef]
- Weyand, C.M.; Brandes, J.C.; Schmidt, D.; Fulbright, J.W.; Goronzy, J.J. Functional properties of CD4+ CD28- T cells in the aging immune system. Mech. Ageing Dev. 1998, 102, 131–147. [Google Scholar] [CrossRef]
- Schirmer, M.; Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J. Immunol. 1998, 161, 1018–1025. [Google Scholar]
- Watad, A.; Bragazzi, N.L.; Adawi, M.; Amital, H.; Toubi, E.; Porat, B.S.; Shoenfeld, Y. Autoimmunity in the Elderly: Insights from Basic Science and Clinics—Mini-Review. Gerontology 2017, 63, 515–523. [Google Scholar] [CrossRef]
- Goyal, R.; Bulua, A.C.; Nikolov, N.P.; Schwartzberg, P.L.; Siegel, R.M. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Curr. Opin. Rheumatol. 2009, 21, 78–84. [Google Scholar] [CrossRef]
- Villa, A.; Marrella, V.; Rucci, F.; Notarangelo, L.D. Genetically determined lymphopenia and autoimmune manifestations. Curr. Opin. Immunol. 2008, 20, 318–324. [Google Scholar] [CrossRef]
- Wada, T.; Toma, T.; Okamoto, H.; Kasahara, Y.; Koizumi, S.; Agematsu, K.; Kimura, H.; Shimada, A.; Hayashi, Y.; Kato, M.; et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood 2005, 106, 2099–2101. [Google Scholar] [CrossRef]
- Park, J.Y.; Kob, M.; Prodeus, A.P.; Rosen, F.S.; Shcherbina, A.; Remold-O’Donnell, E. Early deficit of lymphocytes in Wiskott-Aldrich syndrome: Possible role of WASP in human lymphocyte maturation. Clin. Exp. Immunol. 2004, 136, 104–110. [Google Scholar] [CrossRef]
- Carneiro-Sampaio, M.; Coutinho, A. Tolerance and autoimmunity: Lessons at the bedside of primary immunodeficiencies. Adv. Immunol. 2007, 95, 51–82. [Google Scholar]
- Kaaba, S.A.; Al-Harbi, S.A. Abnormal lymphocyte subsets in Kuwaiti patients with type-1 insulin-dependent diabetes mellitus and their first-degree relatives. Immunol. Lett. 1995, 47, 209–213. [Google Scholar] [CrossRef]
- Hernan, M.A.; Zhang, S.M.; Lipworth, L.; Olek, M.J.; Ascherio, A. Multiple sclerosis and age at infection with common viruses. Epidemiology 2001, 12, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Weetman, A.P. Graves’ disease following immune reconstitution or immunomodulatory treatment: Should we manage it any differently? Clin. Endocrinol. 2014, 80, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Schumacher, M.J. Myasthenia gravis and lymphoma. A clinical and immunological association. JAMA 1979, 242, 2096–2097. [Google Scholar] [CrossRef] [PubMed]
- Dan, D.; Bart, P.A.; Novy, J.; Kuntzer, T.; Clair, C. Double seronegative myasthenia gravis with antiphospholipid syndrome: A case report. J. Med. Case Rep. 2014, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, S.J.; Penhale, W.J.; Irvine, W.J. Circulating lymphocyte subpopulations in Hashimoto thyroiditis. Clin. Exp. Immunol. 1973, 15, 345–354. [Google Scholar]
- Karlsson-Parra, A.; Nyberg, A.; Totterman, T.H.; Loof, L.; Forsum, U. Primary biliary cirrhosis--phenotypic characterization of immunocompetent cells in peripheral blood and liver tissue. Upsala J. Med. Sci. 1984, 89, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Rich, K.C.; Arnold, W.J.; Palella, T.; Fox, I.H. Cellular immune deficiency with autoimmune hemolytic anemia in purine nucleoside phosphorylase deficiency. Am. J. Med. 1979, 67, 172–176. [Google Scholar] [CrossRef]
- Prelog, M.; Schwarzenbrunner, N.; Sailer-Hock, M.; Kern, H.; Klein-Franke, A.; Ausserlechner, M.J.; Koppelstaetter, C.; Brunner, A.; Duftner, C.; Dejaco, C.; et al. Premature aging of the immune system in children with juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 2153–2162. [Google Scholar] [CrossRef]
- Bateman, H.E.; Kirou, K.A.; Paget, S.A.; Crow, M.K.; Yee, A.M. Remission of juvenile rheumatoid arthritis after infection with parvovirus B19. J. Rheumatol. 1999, 26, 2482–2484. [Google Scholar]
- Mirzayan, M.J.; Schmidt, R.E.; Witte, T. Prognostic parameters for flare in systemic lupus erythematosus. Rheumatology 2000, 39, 1316–1319. [Google Scholar] [CrossRef]
- Rivero, S.J.; Diaz-Jouanen, E.; Alarcon-Segovia, D. Lymphopenia in systemic lupus erythematosus. Clinical, diagnostic, and prognostic significance. Arthritis Rheum. 1978, 21, 295–305. [Google Scholar] [CrossRef]
- Utsinger, P.D.; Yount, W.J. Lymphopenia in Sjogren’s syndrome with rheumatoid arthritis: Relationship to lymphocytotoxic antibodies, cryoglobulinemia, and impaired mitogen responsiveness. J. Rheumatol. Int. 1976, 3, 355–366. [Google Scholar]
- Goronzy, J.J.; Weyand, C.M. Aging, autoimmunity and arthritis: T-cell senescence and contraction of T-cell repertoire diversity—catalysts of autoimmunity and chronic inflammation. Arthritis Res. Ther. 2003, 5, 225–234. [Google Scholar] [CrossRef]
- Viguier, M.; Fouere, S.; de la Salmoniere, P.; Rabian, C.; Lebbe, C.; Dubertret, L.; Morel, P.; Bachelez, H. Peripheral blood lymphocyte subset counts in patients with dermatomyositis: Clinical correlations and changes following therapy. Medicine 2003, 82, 82–86. [Google Scholar] [CrossRef]
- Iannone, F.; Cauli, A.; Yanni, G.; Kingsley, G.H.; Isenberg, D.A.; Corrigall, V.; Panayi, G.S. T-lymphocyte immunophenotyping in polymyositis and dermatomyositis. Br. J. Rheumatol. 1996, 35, 839–845. [Google Scholar] [CrossRef][Green Version]
- Kirtava, Z.B.J.; Bredberg, A.; Henriksson, G.; Jacobsson, L.; Manthorpe, R. CD4+ T-lymphocytopenia without HIV infection: Increased prevalence among patients with primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 1995, 13, 609–616. [Google Scholar]
- Czirjak, L.; Danko, K.; Sonkoly, I.; Bodolay, E.; Szegedi, G. Lymphocyte markers in patients with progressive systemic sclerosis. Acta Med. Hung. 1985, 42, 109–114. [Google Scholar]
- Hughes, P.; Holt, S.; Rowell, N.R.; Dodd, J. Thymus-dependent (T) lymphocyte deficiency in progressive systemic sclerosis. Br. J. Dermatol. 1976, 95, 469–473. [Google Scholar] [CrossRef]
- Heimann, T.M.; Bolnick, K.; Aufses, A.H., Jr. Prognostic significance of severe preoperative lymphopenia in patients with Crohn’s disease. Ann. Surg. 1986, 203, 132–135. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science/Taylor & Francis Group, LLC.: New York, NY, USA, 2016; p. 20, 904p. [Google Scholar]
- Miller, F.W.; Lamb, J.A.; Schmidt, J.; Nagaraju, K. Risk factors and disease mechanisms in myositis. Nat. Rev. Rheumatol. 2018, 14, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Fye, K.H.; Terasaki, P.I.; Michalski, J.P.; Daniels, T.E.; Opelz, G.; Talal, N. Relationshipp of HLA-Dw3 and HLA-B8 to Sjogren’s syndrome. Arthritis Rheum. 1978, 21, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C. HLA and autoimmunity in scleroderma (systemic sclerosis). Int. Rev. Immunol. 1995, 12, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Plevy, S.E.; Taylor, K.; Tyan, D.; Fischel-Ghodsian, N.; McElree, C.; Targan, S.R.; Rotter, J.I. Linkage of Crohn’s disease to the major histocompatibility complex region is detected by multiple non-parametric analyses. Gut 1999, 44, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blum, S.S.; Bender, M. The Immune System Recovery Plan: A Doctor’s 4-Step Program to Treat Autoimmune Disease; First Scribner Hardcover Edition; Scribner: New York, NY, USA, 2013; p. 12, 370p. [Google Scholar]
- El-Samahy, M.H.; Tantawy, A.A.G.; Adly, A.A.M.; Habeeb, N.M.; Ismail, E.A.R.; Hamed, G.M.; Eladawy, R. Expression of CD4(+) CD28(null) T lymphocytes in children and adolescents with type 1 diabetes mellitus: Relation to microvascular complications, aortic elastic properties, and carotid intima media thickness. Pediatr. Diabetes 2017, 18, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Thewissen, M.; Linsen, L.; Somers, V.; Geusens, P.; Raus, J.; Stinissen, P. Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann. N. Y. Acad. Sci. 2005, 1051, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Prelog, M.; Schonlaub, J.; Wurzner, R.; Koppelstaetter, C.; Almanzar, G.; Brunner, A.; Gasser, M.; Prommegger, R.; Hausler, G.; Kapelari, K.; et al. Lower CD28+ T cell proportions were associated with CMV-seropositivity in patients with Hashimoto’s thyroiditis. BMC Endocr. Disord. 2013, 13, 34. [Google Scholar] [CrossRef]
- Isse, K.; Harada, K.; Sato, Y.; Nakanuma, Y. Characterization of biliary intra-epithelial lymphocytes at different anatomical levels of intrahepatic bile ducts under normal and pathological conditions: Numbers of CD4+CD28- intra-epithelial lymphocytes are increased in primary biliary cirrhosis. Pathol. Int. 2006, 56, 17–24. [Google Scholar] [CrossRef]
- Youssef, S.R.; Elsalakawy, W.A. First report of expansion of CD4(+)/CD28 null T-helper lymphocytes in adult patients with idiopathic autoimmune hemolytic anemia. Hematol. Transfus. Cell Ther. 2020. [Google Scholar] [CrossRef]
- Kaneko, H.; Saito, K.; Hashimoto, H.; Yagita, H.; Okumura, K.; Azuma, M. Preferential elimination of CD28+ T cells in systemic lupus erythematosus (SLE) and the relation with activation-induced apoptosis. Clin. Exp. Immunol. 1996, 106, 218–229. [Google Scholar] [CrossRef]
- Weyand, C.M.; Fulbright, J.W.; Goronzy, J.J. Immunosenescence, autoimmunity, and rheumatoid arthritis. Exp. Gerontol. 2003, 38, 833–841. [Google Scholar] [CrossRef]
- Fessler, J.; Fasching, P.; Raicht, A.; Hammerl, S.; Weber, J.; Lackner, A.; Hermann, J.; Dejaco, C.; Graninger, W.B.; Schwinger, W.; et al. Lymphopenia in primary Sjogren’s syndrome is associated with premature aging of naive CD4+ T cells. Rheumatology 2020. [Google Scholar] [CrossRef]
- Korman, B. Evolving insights into the cellular and molecular pathogenesis of fibrosis in systemic sclerosis. Transl. Res. 2019, 209, 77–89. [Google Scholar] [CrossRef]
- Howie, H.L.; Hudson, K.E. Murine models of autoimmune hemolytic anemia. Curr. Opin. Hematol. 2018, 25, 473–481. [Google Scholar] [CrossRef]
- Khiong, K.; Murakami, M.; Kitabayashi, C.; Ueda, N.; Sawa, S.; Sakamoto, A.; Kotzin, B.L.; Rozzo, S.J.; Ishihara, K.; Verella-Garcia, M.; et al. Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J. Clin. Investig. 2007, 117, 1270–1281. [Google Scholar] [CrossRef]
- Snapper, S.B.; Rosen, F.S.; Mizoguchi, E.; Cohen, P.; Khan, W.; Liu, C.H.; Hagemann, T.L.; Kwan, S.P.; Ferrini, R.; Davidson, L.; et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 1998, 9, 81–91. [Google Scholar] [CrossRef]
- Zhang, J.; Shehabeldin, A.; da Cruz, L.A.; Butler, J.; Somani, A.K.; McGavin, M.; Kozieradzki, I.; dos Santos, A.O.; Nagy, A.; Grinstein, S.; et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 1999, 190, 1329–1342. [Google Scholar] [CrossRef]
- Fischer, H.J.; Witte, A.K.; Walter, L.; Grone, H.J.; van den Brandt, J.; Reichardt, H.M. Distinct roles of T-cell lymphopenia and the microbial flora for gastrointestinal and CNS autoimmunity. FASEB J. 2016, 30, 1724–1732. [Google Scholar] [CrossRef]
- Penhale, W.J.; Farmer, A.; McKenna, R.P.; Irvine, W.J. Spontaneous thyroiditis in thymectomized and irradiated Wistar rats. Clin. Exp. Immunol. 1973, 15, 225–236. [Google Scholar]
- Jang, E.; Kim, H.R.; Cho, S.H.; Paik, D.J.; Kim, J.M.; Lee, S.K.; Youn, J. Prevention of spontaneous arthritis by inhibiting homeostatic expansion of autoreactive CD4+ T cells in the K/BxN mouse model. Arthritis Rheum. 2006, 54, 492–498. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Grimbacher, B.; Witte, T. Autoimmunity and primary immunodeficiency: Two sides of the same coin? Nat. Rev. Rheumatol. 2017, 14, 7–18. [Google Scholar] [CrossRef]
- Jonkman-Berk, B.M.; van den Berg, J.M.; Ten Berge, I.J.; Bredius, R.G.; Driessen, G.J.; Dalm, V.A.; van Dissel, J.T.; van Deuren, M.; Ellerbroek, P.M.; van der Flier, M.; et al. Primary immunodeficiencies in the Netherlands: National patient data demonstrate the increased risk of malignancy. Clin. Immunol. 2015, 156, 154–162. [Google Scholar] [CrossRef]
- Suarez, F.; Mahlaoui, N.; Canioni, D.; Andriamanga, C.; Dubois d’Enghien, C.; Brousse, N.; Jais, J.P.; Fischer, A.; Hermine, O.; Stoppa-Lyonnet, D. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: A report from the French national registry of primary immune deficiencies. J. Clin. Oncol. 2015, 33, 202–208. [Google Scholar] [CrossRef]
- Shapiro, R.S. Malignancies in the setting of primary immunodeficiency: Implications for hematologists/oncologists. Am. J. Hematol. 2011, 86, 48–55. [Google Scholar] [CrossRef]
- Tuano, K.S.; Orange, J.S.; Sullivan, K.; Cunningham-Rundles, C.; Bonilla, F.A.; Davis, C.M. Food allergy in patients with primary immunodeficiency diseases: Prevalence within the US Immunodeficiency Network (USIDNET). J. Allergy Clin. Immunol. 2015, 135, 273–275. [Google Scholar] [CrossRef][Green Version]
- Saifi, M.; Wysocki, C.A. Autoimmune Disease in Primary Immunodeficiency: At the Crossroads of Anti-Infective Immunity and Self-Tolerance. Immunol. Allergy Clin. N. Am. 2015, 35, 731–752. [Google Scholar] [CrossRef]
- Grimbacher, B.; Warnatz, K.; Yong, P.F.K.; Korganow, A.S.; Peter, H.H. The crossroads of autoimmunity and immunodeficiency: Lessons from polygenic traits and monogenic defects. J. Allergy Clin. Immunol. 2016, 137, 3–17. [Google Scholar] [CrossRef]
- Guffroy, A.; Gies, V.; Martin, M.; Korganow, A.S. Primary immunodeficiency and autoimmunity. Rev. Med. Intern. 2017, 38, 383–392. [Google Scholar] [CrossRef]
- Fischer, A.; Provot, J.; Jais, J.P.; Alcais, A.; Mahlaoui, N.; Members of The Ceredih French Pid Study Group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393. [Google Scholar] [CrossRef]
- Signorini, S.; Imberti, L.; Pirovano, S.; Villa, A.; Facchetti, F.; Ungari, M.; Bozzi, F.; Albertini, A.; Ugazio, A.G.; Vezzoni, P.; et al. Intrathymic restriction and peripheral expansion of the T-cell repertoire in Omenn syndrome. Blood 1999, 94, 3468–3478. [Google Scholar] [CrossRef]
- Gennery, A.R.; Hodges, E.; Williams, A.P.; Harris, S.; Villa, A.; Angus, B.; Cant, A.J.; Smith, J.L. Omenn’s syndrome occurring in patients without mutations in recombination activating genes. Clin. Immunol. 2005, 116, 246–256. [Google Scholar] [CrossRef]
- Villa, A.; Sobacchi, C.; Notarangelo, L.D.; Bozzi, F.; Abinun, M.; Abrahamsen, T.G.; Arkwright, P.D.; Baniyash, M.; Brooks, E.G.; Conley, M.E.; et al. V(D)J recombination defects in lymphocytes due to RAG mutations: Severe immunodeficiency with a spectrum of clinical presentations. Blood 2001, 97, 81–88. [Google Scholar] [CrossRef]
- Marrella, V.; Poliani, P.L.; Casati, A.; Rucci, F.; Frascoli, L.; Gougeon, M.L.; Lemercier, B.; Bosticardo, M.; Ravanini, M.; Battaglia, M.; et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J. Clin. Investig. 2007, 117, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Medioni, J.; Haddad, E.; Quartier, P.; Cavazzana-Calvo, M.; Le Deist, F.; de Saint Basile, G.; Delaunay, J.; Schwarz, K.; Casanova, J.L.; et al. Autoimmunity in Wiskott-Aldrich syndrome: Risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics 2003, 111, e622–e627. [Google Scholar] [CrossRef]
- Imai, K.; Nonoyama, S.; Ochs, H.D. WASP (Wiskott-Aldrich syndrome protein) gene mutations and phenotype. Curr. Opin. Allergy Clin. Immunol. 2003, 3, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Cleland, S.Y.; Siegel, R.M. Wiskott-Aldrich Syndrome at the nexus of autoimmune and primary immunodeficiency diseases. FEBS Lett. 2011, 585, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Adriani, M.; Aoki, J.; Horai, R.; Thornton, A.M.; Konno, A.; Kirby, M.; Anderson, S.M.; Siegel, R.M.; Candotti, F.; Schwartzberg, P.L. Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin. Immunol. 2007, 124, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, F.; Trifari, S.; Scaramuzza, S.; Panaroni, C.; Martino, S.; Notarangelo, L.D.; Baz, Z.; Metin, A.; Cattaneo, F.; Villa, A.; et al. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J. Exp. Med. 2007, 204, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Schurman, S.H.; Candotti, F. Autoimmunity in Wiskott-Aldrich syndrome. Curr. Opin. Rheumatol. 2003, 15, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Pociot, F.; Todd, J.A.; Rich, S.S.; Type 1 Diabetes Genetics Consortium. Type 1 diabetes: Evidence for susceptibility loci from four genome-wide linkage scans in 1435 multiplex families. Diabetes 2005, 54, 2995–3001. [Google Scholar] [CrossRef]
- Krishnan, S.; Chowdhury, B.; Tsokos, G.C. Autoimmunity in systemic lupus erythematosus: Integrating genes and biology. Semin. Immunol. 2006, 18, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Morahan, G.; Morel, L. Genetics of autoimmune diseases in humans and in animal models. Curr. Opin. Immunol. 2002, 14, 803–811. [Google Scholar] [CrossRef]
- Nath, S.K.; Kilpatrick, J.; Harley, J.B. Genetics of human systemic lupus erythematosus: The emerging picture. Curr. Opin. Immunol. 2004, 16, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Tsao, B.P. Update on human systemic lupus erythematosus genetics. Curr. Opin. Rheumatol. 2004, 16, 513–521. [Google Scholar] [CrossRef]
- Wandstrat, A.; Wakeland, E. The genetics of complex autoimmune diseases: Non-MHC susceptibility genes. Nat. Immunol. 2001, 2, 802–809. [Google Scholar] [CrossRef]
- Wicker, L.S.; Clark, J.; Fraser, H.I.; Garner, V.E.; Gonzalez-Munoz, A.; Healy, B.; Howlett, S.; Hunter, K.; Rainbow, D.; Rosa, R.L.; et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J. Autoimmun. 2005, 25, 29–33. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Brown, M.L.; Elenburg, S.N.; Lieberman, J.A.; Michael, C.F.; Srinivasan, S.; Wang, W.C.; Myers, L.K.; Lew, D.B. Autoimmune Features of Wiskott-Aldrich Syndrome: A Case Report. J. Autoimmune Disord. 2016, 2, 1–4. [Google Scholar] [CrossRef][Green Version]
- Shirkani, A.; Farrokhi, S. Evaluation of Classic Wiskott Aldrich Syndrome with Mild Symptoms in Two Cousins: A Case Report. Iran. J. Pediatr. 2017, 27, e5883. [Google Scholar] [CrossRef]
- Stradner, M.H.; Dejaco, C.; Brickmann, K.; Graninger, W.B.; Brezinschek, H.P. A combination of cellular biomarkers predicts failure to respond to rituximab in rheumatoid arthritis: A 24-week observational study. Arthritis Res. Ther. 2016, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.; Nocturne, G.; Cornec, D.; Daien, C.I. Lymphocytes as Biomarkers of Therapeutic Response in Rheumatic Autoimmune Diseases, is it a Realistic Goal? Clin. Rev. Allergy Immunol. 2017, 53, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Khanmohammadi, S.; Shabani, M.; Tabary, M.; Rayzan, E.; Rezaei, N. Lymphoma in the setting of autoimmune diseases: A review of association and mechanisms. Crit. Rev. Oncol. Hematol. 2020, 150, 102945. [Google Scholar] [CrossRef] [PubMed]
- Masciopinto, P.; Dell’Olio, G.; de Robertis, R.; Specchia, G.; Musto, P.; Albano, F. The Role of Autoimmune Diseases in the Prognosis of Lymphoma. J. Clin. Med. 2020, 9, 3403. [Google Scholar] [CrossRef] [PubMed]
- Kurita, D.; Miyoshi, H.; Ichikawa, A.; Kato, K.; Imaizumi, Y.; Seki, R.; Sato, K.; Sasaki, Y.; Kawamoto, K.; Shimono, J.; et al. Methotrexate-associated Lymphoproliferative Disorders in Patients with Rheumatoid Arthritis: Clinicopathologic Features and Prognostic Factors. Am. J. Surg. Pathol. 2019, 43, 869–884. [Google Scholar] [CrossRef]
- Kaji, D.; Kusakabe, M.; Sakata-Yanagimoto, M.; Suehara, Y.; Hattori, K.; Ota, Y.; Yuasa, M.; Kageyama, K.; Taya, Y.; Takagi, S.; et al. Clinicopathological Analysis of “Other Iatrogenic Immunodeficiency-Associated Lymphoproliferative Disorders” Reveals a Favorable Outcome Independent of the Effectiveness of Methotrexate Discontinuation in Autoimmune Disease Patients. Blood 2019, 134 (Suppl. S1), 4128. [Google Scholar] [CrossRef]
- Harigai, M. Lymphoproliferative disorders in patients with rheumatoid arthritis in the era of widespread use of methotrexate: A review of the literature and current perspective. Mod. Rheumatol. 2018, 28, 1–8. [Google Scholar] [CrossRef]
- Yen, Y.F.; Chuang, P.H.; Jen, I.A.; Chen, M.; Lan, Y.C.; Liu, Y.L.; Lee, Y.; Chen, Y.H.; Chen, Y.A. Incidence of autoimmune diseases in a nationwide HIV/AIDS patient cohort in Taiwan, 2000–2012. Ann. Rheum. Dis. 2017, 76, 661–665. [Google Scholar] [CrossRef]
- Grewal, R.; Irimie, A.; Naidoo, N.; Mohamed, N.; Petrushev, B.; Chetty, M.; Tomuleasa, C.; Abayomi, E.A. Hodgkin’s lymphoma and its association with EBV and HIV infection. Crit. Rev. Clin. Lab. Sci. 2018, 55, 102–114. [Google Scholar] [CrossRef]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef]
- Quaresma, J.A.; Yoshikawa, G.T.; Koyama, R.V.; Dias, G.A.; Fujihara, S.; Fuzii, H.T. HTLV-1, Immune Response and Autoimmunity. Viruses 2016, 8, 5. [Google Scholar] [CrossRef]
- Yamagishi, M.; Fujikawa, D.; Watanabe, T.; Uchimaru, K. HTLV-1-Mediated Epigenetic Pathway to Adult T-Cell Leukemia-Lymphoma. Front. Microbiol. 2018, 9, 1686. [Google Scholar] [CrossRef]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Deng, Y.; Weng, Z.; Yang, L. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A systemic review and meta-analysis. Int. J. Infect. Dis. 2020, 96, 131–135. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Bermejo-Martin, J.F.; Almansa, R.; Menendez, R.; Mendez, R.; Kelvin, D.J.; Torres, A. Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J. Infect. 2020, 80, e23–e24. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Ciechanowicz, A.; Sielatycka, K.; Cymer, M.; Marlicz, W.; Kucia, M. SARS-CoV-2 Entry Receptor ACE2 is Expressed on Very Small CD45(-) Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem Cell Rev. Rep. 2020. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Talotta, R.; Robertson, E. Autoimmunity as the comet tail of COVID-19 pandemic. World J. Clin. Cases 2020, 8, 3621–3644. [Google Scholar] [CrossRef]
- Troy, A.E.; Shen, H. Cutting edge: Homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J. Immunol. 2003, 170, 672–676. [Google Scholar] [CrossRef]
- Kieper, W.C.; Burghardt, J.T.; Surh, C.D. A role for TCR affinity in regulating naive T cell homeostasis. J. Immunol. 2004, 172, 40–44. [Google Scholar] [CrossRef]
- Zou, L.; Mendez, F.; Martin-Orozco, N.; Peterson, E.J. Defective positive selection results in T cell lymphopenia and increased autoimmune diabetes in ADAP-deficient BDC2.5-C57BL/6 mice. Eur. J. Immunol. 2008, 38, 986–994. [Google Scholar] [CrossRef]
- Sawa, S.; Kamimura, D.; Jin, G.H.; Morikawa, H.; Kamon, H.; Nishihara, M.; Ishihara, K.; Murakami, M.; Hirano, T. Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 2006, 203, 1459–1470. [Google Scholar] [CrossRef]
- Wiede, F.; La Gruta, N.L.; Tiganis, T. PTPN2 attenuates T-cell lymphopenia-induced proliferation. Nat. Commun. 2014, 5, 3073. [Google Scholar] [CrossRef]
- Calzascia, T.; Pellegrini, M.; Lin, A.; Garza, K.M.; Elford, A.R.; Shahinian, A.; Ohashi, P.S.; Mak, T.W. CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc. Natl. Acad. Sci. USA 2008, 105, 2999–3004. [Google Scholar] [CrossRef]
- Monti, P.; Scirpoli, M.; Maffi, P.; Ghidoli, N.; de Taddeo, F.; Bertuzzi, F.; Piemonti, L.; Falcone, M.; Secchi, A.; Bonifacio, E. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J. Clin. Investig. 2008, 118, 1806–1814. [Google Scholar] [CrossRef]
- Krupica, T., Jr.; Fry, T.J.; Mackall, C.L. Autoimmunity during lymphopenia: A two-hit model. Clin. Immunol. 2006, 120, 121–128. [Google Scholar] [CrossRef]
- Kekalainen, E.; Lehto, M.K.; Smeds, E.; Pontynen, N.; Pekkarinen, P.T.; Ulmanen, I.; Miettinen, A.; Arstila, T.P. Lymphopenia-induced proliferation in the absence of functional Autoimmune regulator (Aire) induces colitis in mice. Immunol. Lett. 2015, 167, 17–22. [Google Scholar] [CrossRef]
- McPherson, S.W.; Heuss, N.D.; Gregerson, D.S. Lymphopenia-induced proliferation is a potent activator for CD4+ T cell-mediated autoimmune disease in the retina. J. Immunol. 2009, 182, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012, 13, 667–673. [Google Scholar] [CrossRef] [PubMed]
- L’Huillier, A.; Ren, G.; Shi, Y.; Zhang, J. A two-hit model of autoimmunity: Lymphopenia and unresponsiveness to TGF-beta signaling. Cell Mol. Immunol. 2012, 9, 369–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wada, T.; Schurman, S.H.; Garabedian, E.K.; Yachie, A.; Candotti, F. Analysis of T-cell repertoire diversity in Wiskott-Aldrich syndrome. Blood 2005, 106, 3895–3897. [Google Scholar] [CrossRef] [PubMed]
- Honig, M.; Schwarz, K. Omenn syndrome: A lack of tolerance on the background of deficient lymphocyte development and maturation. Curr. Opin. Rheumatol. 2006, 18, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Stoll, M.; Schmidt, R.E. Adverse events of desirable gain in immunocompetence: The Immune Restoration Inflammatory Syndromes. Autoimmun. Rev. 2004, 3, 243–249. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Wang, F.; Hou, H.; Luo, Y.; Tang, G.; Wu, S.; Huang, M.; Liu, W.; Zhu, Y.; Lin, Q.; Mao, L.; et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 2020, 5, 10. [Google Scholar] [CrossRef]
- Datta, S.; Sarvetnick, N.E. IL-21 limits peripheral lymphocyte numbers through T cell homeostatic mechanisms. PLoS ONE 2008, 3, e3118. [Google Scholar] [CrossRef]
- Hakim, F.T.; Gress, R.E. Reconstitution of the lymphocyte compartment after lymphocyte depletion: A key issue in clinical immunology. Eur. J. Immunol. 2005, 35, 3099–3102. [Google Scholar] [CrossRef]
- Arnold, B.; Schuler, T.; Hammerling, G.J. Control of peripheral T-lymphocyte tolerance in neonates and adults. Trends Immunol. 2005, 26, 406–411. [Google Scholar] [CrossRef]
- Dujardin, H.C.; Burlen-Defranoux, O.; Boucontet, L.; Vieira, P.; Cumano, A.; Bandeira, A. Regulatory potential and control of Foxp3 expression in newborn CD4+ T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14473–14478. [Google Scholar] [CrossRef]
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594. [Google Scholar] [CrossRef]
- Guerau-de-Arellano, M.; Martinic, M.; Benoist, C.; Mathis, D. Neonatal tolerance revisited: A perinatal window for Aire control of autoimmunity. J. Exp. Med. 2009, 206, 1245–1252. [Google Scholar] [CrossRef]
- Mathis, D.; Benoist, C. A decade of AIRE. Nat. Rev. Immunol. 2007, 7, 645–650. [Google Scholar] [CrossRef]
- Wang, G.; Miyahara, Y.; Guo, Z.; Khattar, M.; Stepkowski, S.M.; Chen, W. “Default” generation of neonatal regulatory T cells. J. Immunol. 2010, 185, 71–78. [Google Scholar] [CrossRef]
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddle, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993, 5, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Alderuccio, F.; Toh, B.H.; Tan, S.S.; Gleeson, P.A.; van Driel, I.R. An autoimmune disease with multiple molecular targets abrogated by the transgenic expression of a single autoantigen in the thymus. J. Exp. Med. 1993, 178, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Paiva, R.S.; Lino, A.C.; Bergman, M.L.; Caramalho, I.; Sousa, A.E.; Zelenay, S.; Demengeot, J. Recent thymic emigrants are the preferential precursors of regulatory T cells differentiated in the periphery. Proc. Natl. Acad. Sci. USA 2013, 110, 6494–6499. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef]
- Attridge, K.; Wang, C.J.; Wardzinski, L.; Kenefeck, R.; Chamberlain, J.L.; Manzotti, C.; Kopf, M.; Walker, L.S. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood 2012, 119, 4656–4664. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef]
- Wan, S.; Xia, C.; Morel, L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J. Immunol. 2007, 178, 271–279. [Google Scholar] [CrossRef]
- Dominitzki, S.; Fantini, M.C.; Neufert, C.; Nikolaev, A.; Galle, P.R.; Scheller, J.; Monteleone, G.; Rose-John, S.; Neurath, M.F.; Becker, C. Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J. Immunol. 2007, 179, 2041–2045. [Google Scholar] [CrossRef]
- Ellestad, K.K.; Lin, J.; Boon, L.; Anderson, C.C. PD-1 Controls Tonic Signaling and Lymphopenia-Induced Proliferation of T Lymphocytes. Front. Immunol. 2017, 8, 1289. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J. Immunol. 2005, 174, 688–695. [Google Scholar] [CrossRef]
- Krieg, C.; Boyman, O.; Fu, Y.X.; Kaye, J. B and T lymphocyte attenuator regulates CD8+ T cell-intrinsic homeostasis and memory cell generation. Nat. Immunol. 2007, 8, 162–171. [Google Scholar] [CrossRef]
- Zhou, Y.; Leng, X.; Mo, C.; Zou, Q.; Liu, Y.; Wang, Y. The p53 effector Perp mediates the persistence of CD4(+) effector memory T-cell undergoing lymphopenia-induced proliferation. Immunol. Lett. 2020, 224, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tumpey, T.M.; Lu, X.; Morken, T.; Zaki, S.R.; Katz, J.M. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 2000, 74, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.S. Influenza A (H5N1) in Hong Kong: An overview. Vaccine 2002, 20 (Suppl. S2), S77–S81. [Google Scholar] [CrossRef]
- Kiepiela, P.; Coovadia, H.M.; Coward, P. T helper cell defect related to severity in measles. Scand. J. Infect. Dis. 1987, 19, 185–192. [Google Scholar] [CrossRef]
- Olson, G.B.; Dent, P.B.; Rawls, W.E.; South, M.A.; Montgomery, J.R.; Melnick, J.L.; Good, R.A. Abnormalities of in vitro lymphocyte responses during rubella virus infections. J. Exp. Med. 1968, 128, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Higgins, P.G.; Davis, L.R.; Willman, J.S.; Jones, S.E.; Kidd, I.M.; Pattison, J.R.; Tyrrell, D.A. Experimental parvoviral infection in humans. J. Infect. Dis. 1985, 152, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Ouyang, J.; Isnard, S.; Lin, J.; Fombuena, B.; Zhu, B.; Routy, J.P. Sharing CD4+ T Cell Loss: When COVID-19 and HIV Collide on Immune System. Front. Immunol. 2020, 11, 596631. [Google Scholar] [CrossRef]
- Getts, M.T.; Miller, S.D. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Triggering of autoimmune diseases by infections. Clin. Exp. Immunol. 2010, 160, 15–21. [Google Scholar] [CrossRef]
- Toussirot, E.; Roudier, J. Epstein-Barr virus in autoimmune diseases. Best Pract Res. Clin. Rheumatol. 2008, 22, 883–896. [Google Scholar] [CrossRef]
- Jog, N.R.; Chakravarty, E.F.; Guthridge, J.M.; James, J.A. Epstein Barr Virus Interleukin 10 Suppresses Anti-inflammatory Phenotype in Human Monocytes. Front. Immunol. 2018, 9, 2198. [Google Scholar] [CrossRef]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef]
- Slobedman, B.; Barry, P.A.; Spencer, J.V.; Avdic, S.; Abendroth, A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J. Virol. 2009, 83, 9618–9629. [Google Scholar] [CrossRef]
- Ayee, R.; Ofori, M.E.O.; Wright, E.; Quaye, O. Epstein Barr Virus Associated Lymphomas and Epithelia Cancers in Humans. J. Cancer 2020, 11, 1737–1750. [Google Scholar] [CrossRef]
- Mbulaiteye, S.M.; Clarke, C.A.; Morton, L.M.; Gibson, T.M.; Pawlish, K.; Weisenburger, D.D.; Lynch, C.F.; Goodman, M.T.; Engels, E.A. Burkitt lymphoma risk in U.S. solid organ transplant recipients. Am. J. Hematol. 2013, 88, 245–250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Human Disorders with Autoimmune Manifestation | Lymphopenia | Disease-Associated Factor | Animal Model with Lymphopenia | The StudyInvolves Marker(s) of Immune Aging | ||
|---|---|---|---|---|---|---|
| Single-Gene Traits [81] | HLA [81] | Virus Infection [86] | ||||
| Primary immunodeficiency disorder | ||||||
| Omenn syndrome | [56,57,58] | RAG1/2, DCLRE1C, LIG4 | [97] | |||
| Wiskott–Aldrich syndrome | [59] | WASP | [98,99] | |||
| Common ADs secondary to a loss of self-tolerance | ||||||
| Organ-specific disorder | ||||||
| Type 1 diabetes | [61] | CTLA-4, INS | DQ2, DQ8, DR3, DR4 | Coxsackievirus B4,CMV, mumps virus, and rubella virus | [7] | [88] c, [47] a,b,c (mouse model) |
| Multiple sclerosis | [62] | DR2 | EBV, measles virus | [100] | [89] a,c | |
| Graves’ disease | [63] | CTLA-4 | DR3 | Coxsackie B virus, retrovirus, and HCV | ||
| Myasthenia gravis | [64,65] | DR3 | HCV, HSV | |||
| Hashimoto’s thyroiditis | [66] | DR5 | HTLV-1, enterovirus, rubella virus, mumps virus, HSV, EBV, and parvovirus | [101] | [90] a,b,c | |
| Primary biliary cirrhosis | [67] | HCV | [91] c | |||
| Autoimmune hemolytic anemia | [68] | EBV, VZV (mouse model) [87] | [92] c | |||
| Systemic disorder | ||||||
| Juvenile idiopathic arthritis | [69] | [69] a,b | ||||
| Juvenile rheumatoid arthritis | [70] | |||||
| Systemic lupus erythematous | [71,72] | C1q | DR3 | EBV | [93] a,c | |
| Rheumatoid arthritis | [73,74] | DR4 | EBV, HCV | [102] | [89] a,c, [94] a,b,c | |
| Dermatomyositis | [75,76] | B*08:01 [82] | ||||
| Polymyositis | [76] | DRB1*03:01 [82] | ||||
| Primary Sjogren’s syndrome | [73,77] | B8, Dw3 [83] | EBV | [95] a,b | ||
| Systemic sclerosis | [78,79] | DQ5, DQ7 [84] | [96] c | |||
| Crohn’s disease | [80] | DQ5 [85] | ||||
| Investigator, Year | Lymphopenia Type | Model, Transferred Donor | Findings a | Reference |
|---|---|---|---|---|
| Le Campion, 2002 | Physiologically-related | Neonate B6 mice, None |
| [24] |
| Min, 2003 | Physiologically-related | Neonate B6 mice, Adult B6 peripheral LN CD4+ T cells |
| [44] |
| Ernst, 1999 | Genetically-induced | TCRα− mice, Whole B6. PL LN cells |
| [4] |
| Min, 2004 | Genetically-induced | TCR transgenic Rag−/− mice, Polyclonal naïve T cells |
| [14] |
| Min, 2005 | Genetically-induced | Rag2−/− (B10.A background) mice, Naïve CD4+ T cells |
| [15] |
| Jang, 2006 | Genetically-induced | K/BxN mice (a cross between KRN TCR transgenic mice on a C57BL/6 background (K/B) and NOD mice), None | Prevention of spontaneous arthritis is mediated by the inhibition of homeostatic expansion of autoreactive CD4+ T cells in the K/BxN mouse model | [102] |
| Khiong, 2007 | Genetically-induced | Rag1 mutation (reduced activity) mice, None | The LIP of CD4+ T cells is involved in the pathogenesis of an Omenn syndrome murine model | [97] |
| Tajima, 2008 | Genetically-induced | Rag2−/− (C57BL/6 background) mice, Naïve CD8+ T cells |
| [13] |
| Zou, 2008 | Genetically-induced | ADAP-deficient mice bred to the BDC2.5 TCR transgenic mice, None |
| [153] |
| Ernst, 1999 | Irradiation | 600 cGy, Whole B6. PL LN cells |
| [4] |
| Sawa, 2006 | Irradiation | 9.5 Gy on a mouse with Gp130 IL-6 receptor mutation (F759 mouse), None |
| [154] |
| Wiede, 2014 | Irradiation | Sub-lethal irradiation (600–650 Gy), PTPN2-deficient naïve CD8+ T cells |
| [155] |
| King, 2004 | Cytokine-mediated | NOD mice, β cell antigen-specific 8.3-NOD CD8+ T cells |
| [7] |
| Calzascia, 2008 | Immunosuppressive cytostatic drug-induced | Cyclophosphamide-induced lymphopenia in RIP-GP mice (a model of beta-islet cell self-reactivity), None |
| [156] |
| Monti, 2008 | Immunosuppressive drugs-induced | Administration of FK506 and rapamycin in islet transplantation patients, None |
| [157] |
| Koetz, 2000 | Impaired thymic function | Rheumatoid arthritis patients,None |
| [49] |
| Krupica, 2006 | Virus infection | HIV-mediated reduction of CD4+ T cells in patients, None |
| [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheu, T.-T.; Chiang, B.-L. Lymphopenia, Lymphopenia-Induced Proliferation, and Autoimmunity. Int. J. Mol. Sci. 2021, 22, 4152. https://doi.org/10.3390/ijms22084152
Sheu T-T, Chiang B-L. Lymphopenia, Lymphopenia-Induced Proliferation, and Autoimmunity. International Journal of Molecular Sciences. 2021; 22(8):4152. https://doi.org/10.3390/ijms22084152
Chicago/Turabian StyleSheu, Ting-Ting, and Bor-Luen Chiang. 2021. "Lymphopenia, Lymphopenia-Induced Proliferation, and Autoimmunity" International Journal of Molecular Sciences 22, no. 8: 4152. https://doi.org/10.3390/ijms22084152
APA StyleSheu, T.-T., & Chiang, B.-L. (2021). Lymphopenia, Lymphopenia-Induced Proliferation, and Autoimmunity. International Journal of Molecular Sciences, 22(8), 4152. https://doi.org/10.3390/ijms22084152