
Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology





Abstract
1. Introduction
2. Results
2.1. CpG Sites Identified and Individual Correlations with Age
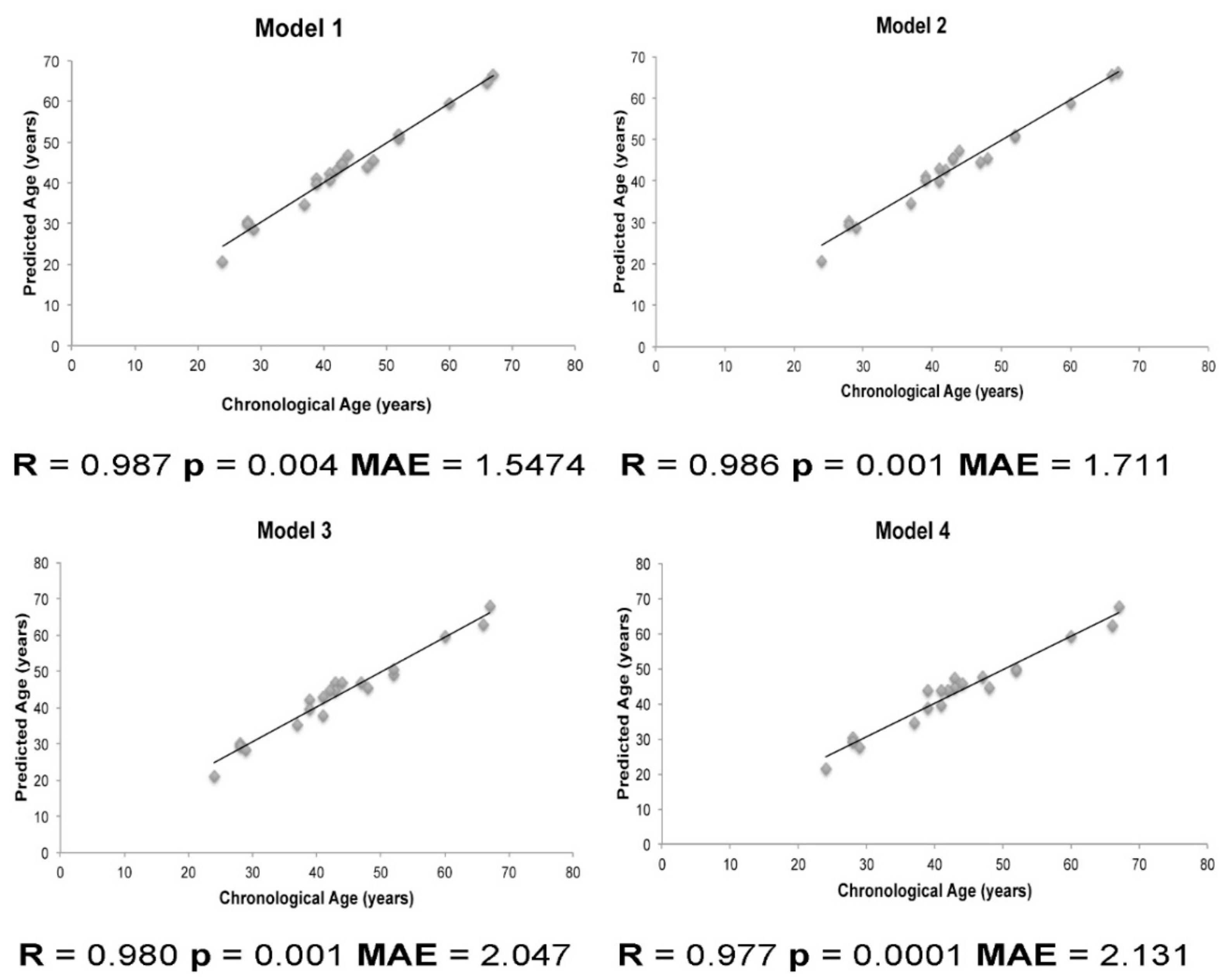
2.2. Construction of Prediction Models for Age Estimation
3. Discussion
4. Material and Methods
4.1. Sample Collection and Teeth Processing
4.2. DNA Extraction
4.3. DNA Quantification
4.4. Bisulfite Conversion and PCR
4.5. Pyrosequencing
4.6. Methylation Results Analyses and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cunha, E.; Baccino, E.; Martrille, L.; Ramsthaler, F.; Prieto, J.; Schuliar, Y.; Lynnerup, N.; Cattaneo, C. The problem of aging human remains and living individuals: A review. Forensic Sci. Int. 2009, 193, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ubelaker, D.H. Estimating age at death from immature human skeletons: An overview. J. Forensic Sci. 1987, 32, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.M.; Maples, W.R. Stages of epiphyseal union for thoracic and lumbar vertebral centra as a method of age determination for teenage and young adult skeletons. J. Forensic Sci. 1995, 40, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Lynnerup, N.; Belard, E.; Buch-Olsen, K.; Sejrsen, B.; Damgaard-Pedersen, K. Intra- and interobserver error of the Greulich-Pyle method as used on a Danish forensic sample. Forensic Sci. Int. 2008, 179, 242.e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Pfau, R.O.; Sciulli, P.W. A method for establishing the age of subadults. J. Forensic Sci. 1994, 39, 165–176. [Google Scholar] [CrossRef]
- Demirjian, A. Tooth eruption in the French Canadian child. J. Dent. Que 1973, 10, 9. [Google Scholar] [PubMed]
- Gustafson, G.; Koch, G. Age estimation up to 16 years of age based on dental development. Odontol. Rev. 1974, 25, 297–306. [Google Scholar]
- Lamendin, H.; Baccino, E.; Humbert, J.F.; Tavernier, J.C.; Nossintchouk, R.M.; Zerilli, A. A simple technique for age estimation in adult corpses: The two criteria dental method. J. Forensic Sci. 1992, 37, 1373–1379. [Google Scholar] [CrossRef]
- Brooks, S.T. Skeletal age at death: The reliability of cranial and pubic age indicators. Am. J. Phys. Anthropol. 1955, 13, 567–597. [Google Scholar] [CrossRef]
- Lovejoy, C.O.; Meindl, R.S.; Pryzbeck, T.R.; Mensforth, R.P. Chronological metamorphosis of the auricular surface of the ilium: A new method for the determination of adult skeletal age at death. Am. J. Phys. Anthropol. 1985, 68, 15–28. [Google Scholar] [CrossRef]
- Iscan, M.Y.; Loth, S.R.; Wright, R.K. Age estimation from the rib by phase analysis: White males. J. Forensic Sci. 1984, 29, 1094–1104. [Google Scholar]
- Meissner, C.; Ritz-Timme, S. Molecular pathology and age estimation. Forensic Sci. Int. 2010, 203, 34–43. [Google Scholar] [CrossRef]
- Ohtani, S.; Utsunomiya, J.; Minoshima, T.; Yamamoto, K. Tooth-based age estimation of an adipocerated cadaver using the amino acid racemization method. Nihon Hoigaku Zasshi Jpn. J. Leg. Med. 1994, 48, 279–281. [Google Scholar]
- Ohtani, S.; Yamamoto, T. Strategy for the estimation of chronological age using the aspartic acid racemization method with special reference to coefficient of correlation between D/L ratios and ages. J. Forensic Sci. 2005, 50, 1020–1027. [Google Scholar] [CrossRef]
- Dobberstein, R.C.; Huppertz, J.; von Wurmb-Schwark, N.; Ritz-Timme, S. Degradation of biomolecules in artificially and naturally aged teeth: Implications for age estimation based on aspartic acid racemization and DNA analysis. Forensic Sci. Int. 2008, 179, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Sirin, N.; Matzenauer, C.; Reckert, A.; Ritz-Timme, S. Age estimation based on aspartic acid racemization in dentine: What about caries-affected teeth? Int. J. Leg. Med. 2018, 132, 623–628. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.; Katari, S.; Coutifaris, C.; Sapienza, C. Explaining inter-individual variability in phenotype: Is epigenetics up to the challenge? Epigenetics 2010, 5, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Declerck, K.; Vanden Berghe, W. Back to the future: Epigenetic clock plasticity towards healthy aging. Mech. Ageing Dev. 2018, 174, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome. Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Silva, D.; Antunes, J.; Balamurugan, K.; Duncan, G.; Alho, C.S.; McCord, B. Developmental validation studies of epigenetic DNA methylation markers for the detection of blood, semen and saliva samples. Forensic Sci. Int. Genet. 2016, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef]
- Hong, S.R.; Jung, S.E.; Lee, E.H.; Shin, K.J.; Yang, W.I.; Lee, H.Y. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017, 29, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Correia Dias, H.; Cordeiro, C.; Corte Real, F.; Cunha, E.; Manco, L. Age Estimation Based on DNA Methylation Using Blood Samples From Deceased Individuals. J. Forensic Sci. 2020, 65, 465–470. [Google Scholar] [CrossRef]
- Giuliani, C.; Cilli, E.; Bacalini, M.G.; Pirazzini, C.; Sazzini, M.; Gruppioni, G.; Franceschi, C.; Garagnani, P.; Luiselli, D. Inferring chronological age from DNA methylation patterns of human teeth. Am. J. Phys. Anthropol. 2016, 159, 585–595. [Google Scholar] [CrossRef]
- Marquez-Ruiz, A.B.; Gonzalez-Herrera, L.; Luna, J.D.; Valenzuela, A. DNA methylation levels and telomere length in human teeth: Usefulness for age estimation. Int. J. Leg. Med. 2020, 134, 451–459. [Google Scholar] [CrossRef]
- Correia Dias, H.; Corte-Real, F.; Cunha, E.; Manco, L. DNA methylation age estimation from human bone and teeth. Aust. J. Forensic Sci. 2020. [Google Scholar] [CrossRef]
- Sweet, D.J.; Sweet, C.H. DNA analysis of dental pulp to link incinerated remains of homicide victim to crime scene. J. Forensic Sci. 1995, 40, 310–314. [Google Scholar] [CrossRef]
- Veeraraghavan, G.; Lingappa, A.; Shankara, S.P.; Mamatha, G.P.; Sebastian, B.T.; Mujib, A. Determination of sex from tooth pulp tissue. Libyan J. Med. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Potsch, L.; Meyer, U.; Rothschild, S.; Schneider, P.M.; Rittner, C. Application of DNA techniques for identification using human dental pulp as a source of DNA. Int. J. Leg. Med. 1992, 105, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.E.; Kelder, B.; Bobik, E.G.; Chuang, L.T.; Lewis, C.J.; Kopchick, J.J.; Mukerji, P.; Huang, Y.S. Identification and expression of mammalian long-chain PUFA elongation enzymes. Lipids 2002, 37, 733–740. [Google Scholar] [CrossRef]
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Giron-Santamaria, L.; Gomez-Tato, A.; de Cal, M.C.; Alvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Espanol, M.; Schneider, P.M.; et al. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016, 24, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Hoefsloot, H.C.J.; Mook, O.R.F.; Rijlaarsdam-Hoekstra, L.; van der Zwalm, M.C.H.; Henneman, P.; Kloosterman, A.D.; Verschure, P.J. Chronological age prediction based on DNA methylation: Massive parallel sequencing and random forest regression. Forensic Sci. Int. Genet. 2017, 31, 19–28. [Google Scholar] [CrossRef]
- Naue, J.; Sanger, T.; Hoefsloot, H.C.J.; Lutz-Bonengel, S.; Kloosterman, A.D.; Verschure, P.J. Proof of concept study of age-dependent DNA methylation markers across different tissues by massive parallel sequencing. Forensic Sci. Int. Genet. 2018, 36, 152–159. [Google Scholar] [CrossRef]
- Dias, H.C.; Cordeiro, C.; Pereira, J.; Pinto, C.; Real, F.C.; Cunha, E.; Manco, L. DNA methylation age estimation in blood samples of living and deceased individuals using a multiplex SNaPshot assay. Forensic Sci. Int. 2020, 311, 110267. [Google Scholar] [CrossRef]
- Zbiec-Piekarska, R.; Spolnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Z.; Paleczka, A.; Kucharczyk, K.; Ploski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef]
- Zbiec-Piekarska, R.; Spolnicka, M.; Kupiec, T.; Makowska, Z.; Spas, A.; Parys-Proszek, A.; Kucharczyk, K.; Ploski, R.; Branicki, W. Examination of DNA methylation status of the ELOVL2 marker may be useful for human age prediction in forensic science. Forensic Sci. Int. Genet. 2015, 14, 161–167. [Google Scholar] [CrossRef]
- Canault, M.; Tellier, E.; Bonardo, B.; Mas, E.; Aumailley, M.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. FHL2 interacts with both ADAM-17 and the cytoskeleton and regulates ADAM-17 localization and activity. J. Cell. Physiol. 2006, 208, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, F.W.; Reischmann, S.; Schwalm, A.; Unger, A.; Ramanujam, D.; Munch, J.; Muller, O.J.; Hengstenberg, C.; Galve, E.; Charron, P.; et al. FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 2014, 109, 451. [Google Scholar] [CrossRef]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.Y.; Richards, H.B.; Consortium, G.; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef]
- Alghanim, H.; Antunes, J.; Silva, D.; Alho, C.S.; Balamurugan, K.; McCord, B. Detection and evaluation of DNA methylation markers found at SCGN and KLF14 loci to estimate human age. Forensic Sci. Int. Genet. 2017, 31, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Skovhus, K.V.; Bergholdt, R.; Erichsen, C.; Sparre, T.; Nerup, J.; Karlsen, A.E.; Pociot, F. Identification and characterization of secretagogin promoter activity. Scand. J. Immunol. 2006, 64, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.B.; Hickey, L.; Michael, G.J.; Derkacs, M.; Christian, L.M.; Kalaitzakis, M.E.; Pearce, R.K.; Graeber, M.B. Neuronal pentraxin II is highly upregulated in Parkinson’s disease and a novel component of Lewy bodies. Acta Neuropathol. 2008, 115, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Soares Bispo Santos Silva, D.; Antunes, J.; Balamurugan, K.; Duncan, G.; Sampaio Alho, C.; McCord, B. Evaluation of DNA methylation markers and their potential to predict human aging. Electrophoresis 2015, 36, 1775–1780. [Google Scholar] [CrossRef]
- Zapico, S.C.; Ubelaker, D.H.; Adserias-Garriga, J. Applications of physiological bases of ageing to forensic sciences. Estimation of age-at-death. Ageing Res. Rev. 2013, 12, 605–617. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Site | r | p-Value |
|---|---|---|---|
| ELOVL2 | CpG1 | 0.353 | 0.024 |
| CpG2 | 0.308 | 0.043 | |
| CpG3 | 0.341 | 0.028 | |
| CpG4 | 0.318 | 0.038 | |
| CpG5 | 0.240 | 0.093 | |
| CpG6 | 0.342 | 0.028 | |
| CpG7 | 0.365 | 0.020 | |
| KLF14 | CpG1 | 0.168 | 0.480 |
| CpG2 | 0.316 | 0.174 | |
| CpG3 | 0.154 | 0.518 | |
| CpG4 | 0.278 | 0.236 | |
| CpG5 | 0.267 | 0.256 | |
| CpG6 | 0.220 | 0.350 | |
| CpG7 | 0.468 | 0.037 | |
| SCGN | CpG1 | 0.313 | 0.180 |
| CpG2 | 0.344 | 0.138 | |
| CpG3 | 0.529 | 0.017 | |
| CpG4 | 0.340 | 0.142 | |
| CpG5 | −0.103 | 0.665 | |
| CpG6 | 0.268 | 0.254 | |
| CpG7 | 0.258 | 0.272 | |
| CpG8 | 0.508 | 0.022 | |
| CpG9 | 0.156 | 0.512 | |
| CpG10 | 0.291 | 0.213 | |
| NPTX2 | CpG1 | 0.280 | 0.076 |
| CpG2 | −0.105 | 0.514 | |
| CpG3 | −0.084 | 0.601 | |
| CpG4 | 0.327 | 0.037 | |
| CpG5 | 0.214 | 0.179 | |
| CpG6 | 0.022 | 0.890 | |
| CpG7 | 0.121 | 0.449 | |
| CpG8 | 0.151 | 0.346 | |
| CpG9 | 0.076 | 0.637 | |
| CpG10 | 0.136 | 0.396 | |
| CpG11 | 0.172 | 0.282 | |
| CpG12 | 0.127 | 0.428 | |
| CpG13 | 0.166 | 0.300 | |
| CpG14 | 0.136 | 0.556 | |
| FHL2 | CpG1 | −0.367 | 0.111 |
| CpG2 | −0.376 | 0.094 | |
| CpG3 | −0.251 | 0.285 | |
| CpG4 | −0.288 | 0.217 | |
| CpG5 | −0.262 | 0.264 | |
| CpG6 | −0.241 | 0.305 | |
| CpG7 | −0.088 | 0.713 | |
| CpG8 | −0.086 | 0.720 |
| Model | R | R2 | SE | p-Value | MAE | MAE (LOOCV) |
|---|---|---|---|---|---|---|
| Age (years) = 12.763 + 4.034(ELOVL2CpG3) + 3.535(ELOVL2CpG4) − 3.040(ELOVL2CpG7) + 7.815(NPTX2CpG4) + 3.791(SCGNCpG3) + 9.122(SCGNCpG8) − 5.013(ELOVL2CpG2) − 1.643(ELOVL2CpG5) − 4.341(FHL2CpG1) + 3.571(FHL2CpG3) − 1.093(FHL2CpG4) + 3.882(FHL2CpG5) − 1.229(FHL2CpG6) − 1.662(KLF14CpG7) | 0.987 | 0.975 | 3.671 | 0.004 | 1.5474 | 2.128 |
| Age (years) = 14.710 + 3.675(ELOVL2CpG3) + 3.972(ELOVL2CpG4) − 2.978(ELOVL2CpG7) + 5.278(NPTX2CpG4) + 4.044(SCGNCpG3) + 8.378(SCGNCpG8) − 4.853(ELOVL2CpG2) − 1.875(ELOVL2CpG5) − 4.273(FHL2CpG1) + 3.547(FHL2CpG3) − 1.145(FHL2CpG4) + 3.640(FHL2CpG5) − 0.937(FHL2CpG6) | 0.986 | 0.972 | 3.505 | 0.001 | 1.711 | 1.706 |
| Age (years) = 14.349 + 4.635(ELOVL2CpG3) + 3.049(ELOVL2CpG4) − 3.681(ELOVL2CpG7) + 5.254(NPTX2CpG4) + 3.810(SCGNCpG3) + 9.503(SCGNCpG8) − 4.835(ELOVL2CpG2) − 0.982(ELOVL2CpG5) − 4.191(FHL2CpG1) + 3.778(FHL2CpG3) − 1.447(FHL2CpG4) + 2.638(FHL2CpG5) | 0.980 | 0.961 | 3.874 | 0.001 | 2.047 | 2.083 |
| Age (years) = 14.854 + 5.139(ELOVL2CpG3) + 2.249(ELOVL2CpG4) − 4.086(ELOVL2CpG7) + 6.927(NPTX2CpG4) + 3.505(SCGNCpG3) + 10.363(SCGNCpG8) − 4.983(ELOVL2CpG2) − 4.223(FHL2CpG1) + 4.075(FHL2CpG3) − 1.562(FHL2CpG4) + 2.506(FHL2CpG5) | 0.977 | 0.955 | 3.866 | 0.0001 | 2.1313 | 1.942 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
C. Zapico, S.; Gauthier, Q.; Antevska, A.; McCord, B.R. Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology. Int. J. Mol. Sci. 2021, 22, 3717. https://doi.org/10.3390/ijms22073717
C. Zapico S, Gauthier Q, Antevska A, McCord BR. Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology. International Journal of Molecular Sciences. 2021; 22(7):3717. https://doi.org/10.3390/ijms22073717
Chicago/Turabian StyleC. Zapico, Sara, Quentin Gauthier, Aleksandra Antevska, and Bruce R. McCord. 2021. "Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology" International Journal of Molecular Sciences 22, no. 7: 3717. https://doi.org/10.3390/ijms22073717
APA StyleC. Zapico, S., Gauthier, Q., Antevska, A., & McCord, B. R. (2021). Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology. International Journal of Molecular Sciences, 22(7), 3717. https://doi.org/10.3390/ijms22073717