
Tracing dsDNA Virus–Host Coevolution through Correlation of Their G-Quadruplex-Forming Sequences

 ,
,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
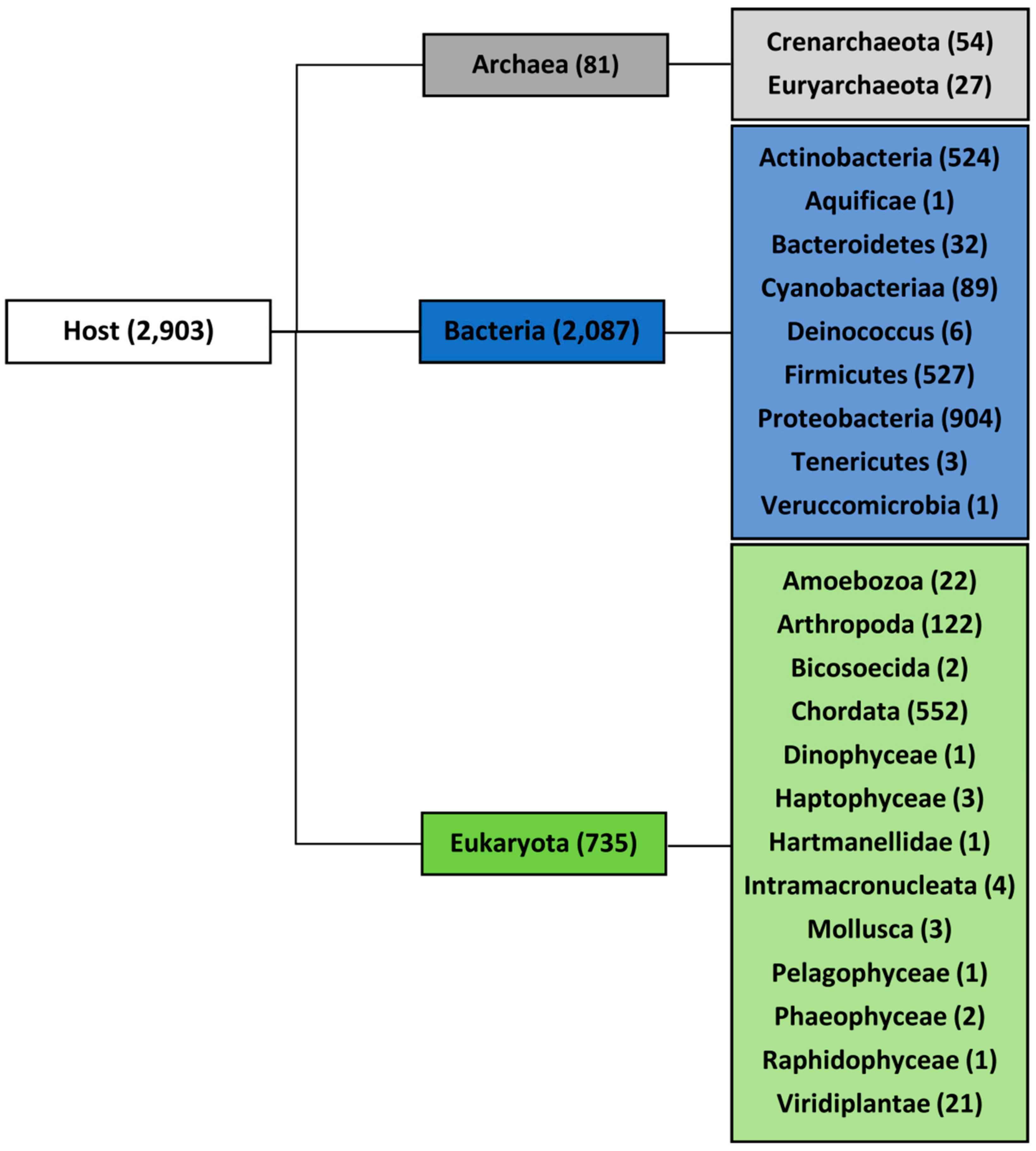
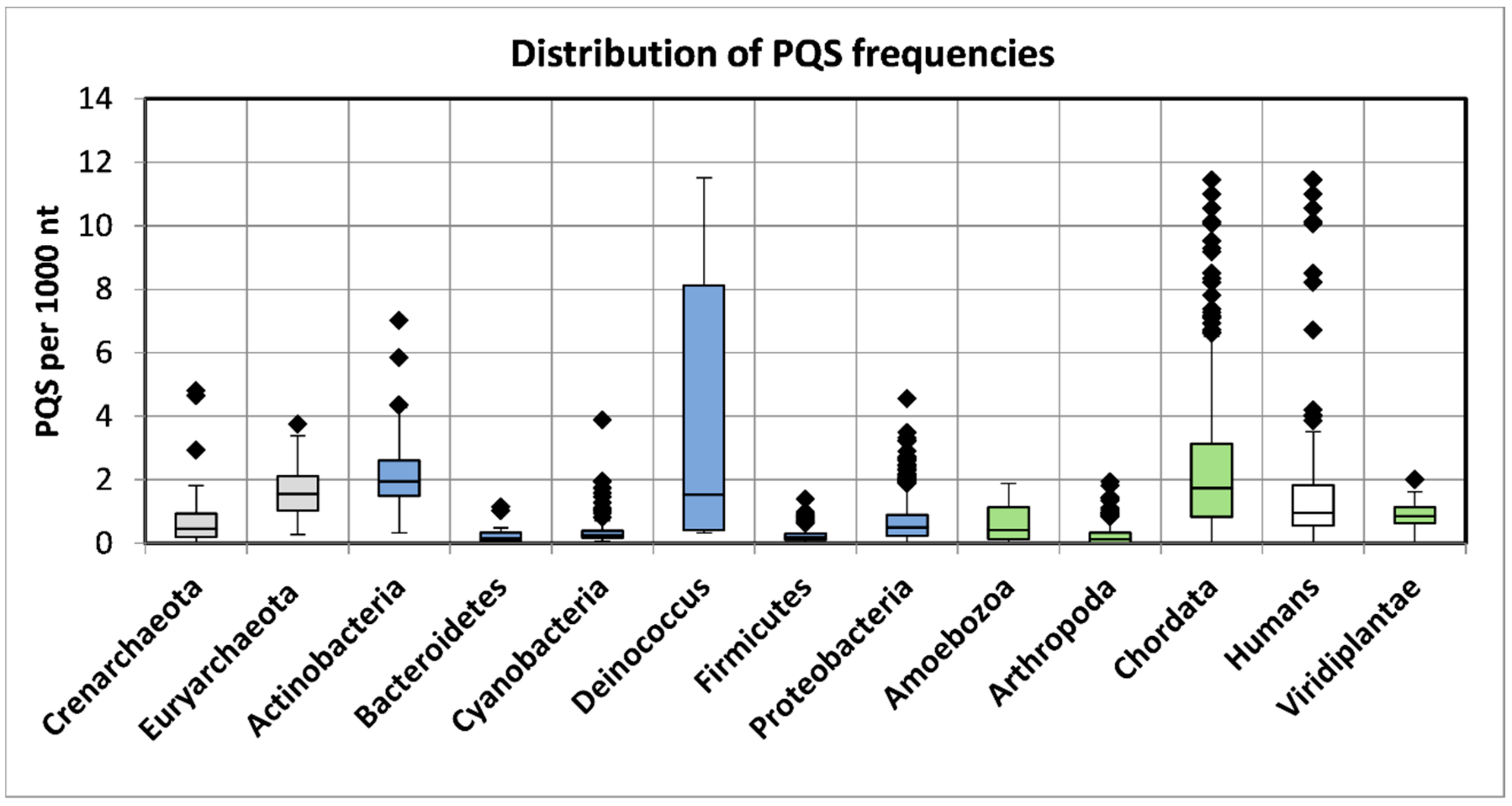
2.1. Variation in Frequency for G4-Forming Sequences in dsDNA Viruses Grouped by Host
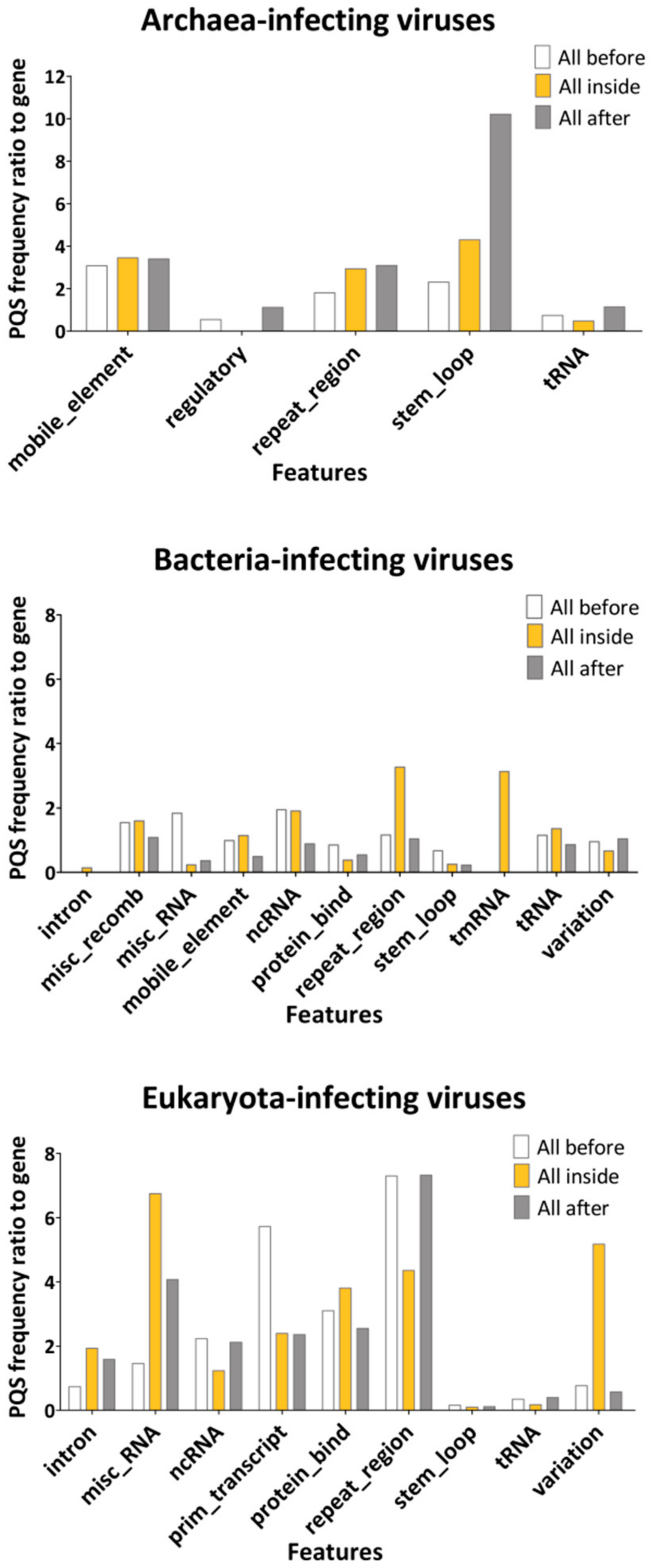
2.2. Features Characteristic for Hosts Are Enriched for PQS in Corresponding dsDNA Viral Genomes
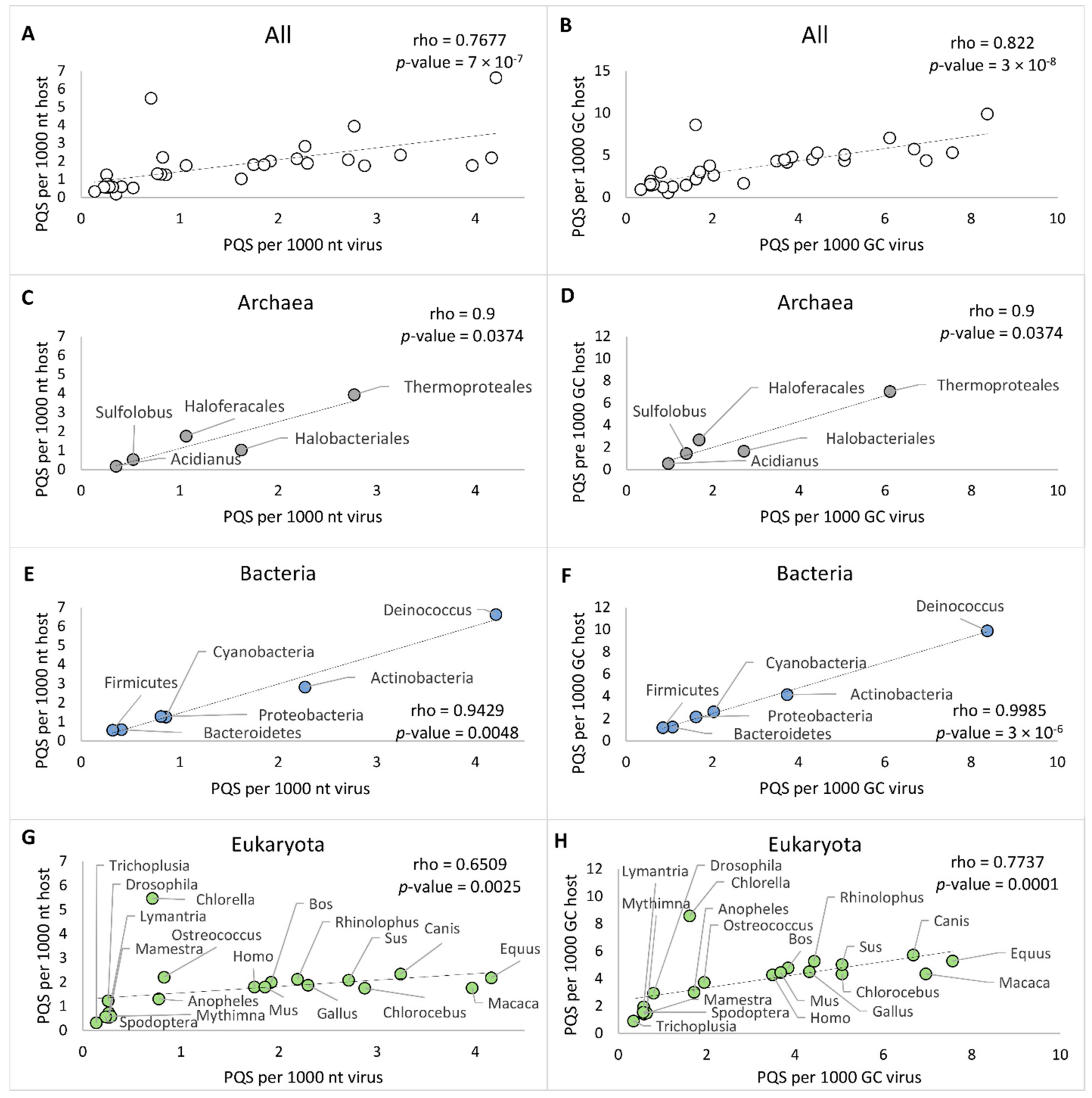
2.3. PQS Frequencies of dsDNA Viruses Correlate with Their Hosts’ Genomes
3. Materials and Methods
3.1. Viral and Host Sequences
3.2. PQS Analyses
3.3. Statistical Analysis
3.4. Overlay of PQS with Annotated Features from NCBI
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EBV | Epstein–Barr virus |
| G4s | G-quadruplexes |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HIV | human immunodeficiency virus |
| KSHV | Kaposi sarcoma herpes virus |
| PQS | putative G-quadruplex-forming sequences |
| NCBI | National Center for Biotechnology Information |
References
- McLaughlin, R.N.; Malik, H.S. Genetic conflicts: The usual suspects and beyond. J. Exp. Biol. 2017, 220, 6–17. [Google Scholar] [CrossRef]
- Kaján, G.L.; Doszpoly, A.; Tarján, Z.L.; Vidovszky, M.Z.; Papp, T. Virus–Host Coevolution with a Focus on Animal and Human DNA Viruses. J. Mol. Evol. 2020, 88, 41–56. [Google Scholar] [CrossRef]
- Charpentier, E.; Doudna, J.A. Rewriting a genome. Nat. Cell Biol. 2013, 495, 50–51. [Google Scholar] [CrossRef]
- Moelling, K.; Broecker, F.; Russo, G.; Sunagawa, S. RNase H as Gene Modifier, Driver of Evolution and Antiviral Defense. Front. Microbiol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.J.; Webster, J.P.; Domingo, E.; Charlesworth, B.; Levin, B.R. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet. 2002, 32, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Lemarteleur, T.; Gomez, D.; Paterski, R.; Mandine, E.; Mailliet, P.; Riou, J.-F. Stabilization of the c-myc gene promoter quadruplex by specific ligands’ inhibitors of telomerase. Biochem. Biophys. Res. Commun. 2004, 323, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5’-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Tawani, A.; Mishra, A.; Kumar, A. G4IPDB: A database for G-quadruplex structure forming nucleic acid interacting proteins. Sci. Rep. 2016, 6, 38144. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Hároníková, L.; Liao, J.C.C.; Fojta, M. DNA and RNA Quadruplex-Binding Proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed]
- Alavi, S.; Ghadiri, H.; Dabirmanesh, B.; Moriyama, K.; Khajeh, K.; Masai, H. G-quadruplex binding protein Rif1, a key regulator of replication timing. J. Biochem. 2021, 169, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Červeň, J.; Bartas, M.; Mikysková, N.; Coufal, J.; Pečinka, P. The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors. Molecules 2018, 23, 2341. [Google Scholar] [CrossRef]
- Masuzawa, T.; Oyoshi, T. Roles of the RGG Domain and RNA Recognition Motif of Nucleolin in G-Quadruplex Stabilization. ACS Omega 2020, 5, 5202–5208. [Google Scholar] [CrossRef]
- Brázda, V.; Coufal, J.; Liao, J.C.; Arrowsmith, C.H. Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem. Biophys. Res. Commun. 2012, 422, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Tosoni, E.; Frasson, I.; Scalabrin, M.; Perrone, R.; Butovskaya, E.; Nadai, M.; Palù, G.; Fabris, D.; Richter, S.N. Nucleolin stabilizes G-quadruplex structures folded by the LTR promoter and silences HIV-1 viral transcription. Nucleic Acids Res. 2015, 43, 8884–8897. [Google Scholar] [CrossRef] [PubMed]
- Bartas, M.; Brázda, V.; Bohálová, N.; Cantara, A.; Volná, A.; Stachurová, T.; Malachová, K.; Jagelská, E.B.; Porubiaková, O.; Červeň, J.; et al. In-Depth Bioinformatic Analyses of Nidovirales Including Human SARS-CoV-2, SARS-CoV, MERS-CoV Viruses Suggest Important Roles of Non-canonical Nucleic Acid Structures in Their Lifecycles. Front. Microbiol. 2020, 11, 1583. [Google Scholar] [CrossRef]
- Kusov, Y.; Tan, J.; Alvarez, E.; Enjuanes, L.; Hilgenfeld, R. A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication–transcription complex. Virology 2015, 484, 313–322. [Google Scholar] [CrossRef]
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Case studies on potential G-quadruplex-forming sequences from the bacterial orders Deinococcales and Thermales derived from a survey of published genomes. Sci. Rep. 2018, 8, 15679. [Google Scholar] [CrossRef]
- Bartas, M.; Čutová, M.; Brázda, V.; Kaura, P.; Šťastný, J.; Kolomazník, J.; Coufal, J.; Goswami, P.; Červeň, J.; Pečinka, P. The Presence and Localization of G-Quadruplex Forming Sequences in the Domain of Bacteria. Molecules 2019, 24, 1711. [Google Scholar] [CrossRef]
- Brazda, V.; Fojta, M.; Bowater, R.P. Structures and stability of simple DNA repeats from bacteria. Biochem. J. 2020, 477, 325–339. [Google Scholar] [CrossRef]
- Ruggiero, E.; Richter, S.N. Viral G-quadruplexes: New frontiers in virus pathogenesis and antiviral therapy. Annu. Rep. Med. Chem. 2020, 54, 101–131. [Google Scholar] [CrossRef] [PubMed]
- Saranathan, N.; Vivekanandan, P. G-Quadruplexes: More Than Just a Kink in Microbial Genomes. Trends Microbiol. 2019, 27, 148–163. [Google Scholar] [CrossRef]
- Wang, S.-R.; Zhang, Q.-Y.; Wang, J.-Q.; Ge, X.-Y.; Song, Y.-Y.; Wang, Y.-F.; Li, X.-D.; Fu, B.-S.; Xu, G.-H.; Shu, B.; et al. Chemical Targeting of a G-Quadruplex RNA in the Ebola Virus L Gene. Cell Chem. Biol. 2016, 23, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Jaubert, C.; Bedrat, A.; Bartolucci, L.; Di Primo, C.; Ventura, M.; Mergny, J.-L.; Amrane, S.; Andreola, M.-L. RNA synthesis is modulated by G-quadruplex formation in Hepatitis C virus negative RNA strand. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Frasson, I.; Nadai, M.; Richter, S.N. Conserved G-Quadruplexes Regulate the Immediate Early Promoters of Human Alphaherpesviruses. Molecules 2019, 24, 2375. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Le, C.; Tyrrell, D.L.; Le, X.C.; Li, X.-F. Aptamer Binding Assay for the E Antigen of Hepatitis B Using Modified Aptamers with G-Quadruplex Structures. Anal. Chem. 2020, 92, 6495–6501. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-quadruplexes regulate Epstein-Barr virus–encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef]
- Dabral, P.; Babu, J.; Zareie, A.; Verma, S.C. LANA and hnRNP A1 Regulate the Translation of LANA mRNA through G-Quadruplexes. J. Virol. 2020, 94, 94. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Richter, S.N. G-quadruplexes and G-quadruplex ligands: Targets and tools in antiviral therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef]
- De Cian, A.; Gros, J.; Guédin, A.; Haddi, M.; Lyonnais, S.; Guittat, L.; Riou, J.-F.; Trentesaux, C.; Saccà, B.; Lacroix, L.; et al. DNA and RNA Quadruplex ligands. Nucleic Acids Symp. Ser. 2008, 52, 7–8. [Google Scholar] [CrossRef]
- Lombardi, E.P.P.; Londoño-Vallejo, A.; Nicolas, A. Relationship Between G-Quadruplex Sequence Composition in Viruses and Their Hosts. Molecules 2019, 24, 1942. [Google Scholar] [CrossRef]
- Brázda, V.; Kolomazník, J.; Lýsek, J.; Bartas, M.; Fojta, M.; Šťastný, J.; Mergny, J.-L. G4Hunter web application: A web server for G-quadruplex prediction. Bioinformatics 2019, 35, 3493–3495. [Google Scholar] [CrossRef]
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, E.P.; Londoño-Vallejo, A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020, 48, 1–15. [Google Scholar] [CrossRef]
- Lightfoot, H.L.; Hagen, T.; Tatum, N.J.; Hall, J. The diverse structural landscape of quadruplexes. FEBS Lett. 2019, 593, 2083–2102. [Google Scholar] [CrossRef]
- Guédin, A.; Gros, J.; Alberti, P.; Mergny, J.-L. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010, 38, 7858–7868. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Ofir, G.; Sorek, R. Contemporary Phage Biology: From Classic Models to New Insights. Cell 2018, 172, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Dunigan, D.D. Chloroviruses: Not your everyday plant virus. Trends Plant. Sci. 2012, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hull, R. Comparative Plant. Virology, 2nd ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Boston, MA, USA, 2009; ISBN 9780123741547. [Google Scholar]
- Marcel, V.; Tran, P.L.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.-P.; Hall, J.; Mergny, J.-L.; Hainaut, P.; Van Dyck, E. G-quadruplex structures in TP53 intron 3: Role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 2010, 32, 271–278. [Google Scholar] [CrossRef]
- Brázda, V.; Luo, Y.; Bartas, M.; Kaura, P.; Porubiaková, O.; Šťastný, J.; Pečinka, P.; Verga, D.; Da Cunha, V.; Takahashi, T.S.; et al. G-Quadruplexes in the Archaea Domain. Biomolecules 2020, 10, 1349. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Kolomazník, J.; Lýsek, J.; Hároníková, L.; Coufal, J.; Št’Astný, J. Palindrome analyser—A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem. Biophys. Res. Commun. 2016, 478, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, P.; Chowdhury, S. QuadBase2: Web server for multiplexed guanine quadruplex mining and visualization. Nucleic Acids Res. 2016, 44, 277–283. [Google Scholar] [CrossRef] [PubMed][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G4Hunter Score | PQS Frequency per 1000 nt | |||
|---|---|---|---|---|
| All | Archaea | Bacteria | Eukaryota | |
| 1.2–1.4 | 1.27 | 1.74 | 0.88 | 1.46 |
| 1.4–1.6 | 0.039 | 0.025 | 0.026 | 0.047 |
| 1.6–1.8 | 0.0042 | 0 | 0.00088 | 0.0062 |
| 1.8–2.0 | 0.00025 | 0 | 0.000041 | 0.00038 |
| 2.0 and more | 0.00021 | 0 | 0.000050 | 0.00031 |
| All | Seq | Median | GC% | PQS | Mean f | Min f | Max f | Cov |
| All | 3134 | 44,746.5 | 44.94 | 220,569 | 1.32 | 0 | 11.51 | 3.34 |
| Domain | Seq | Median | GC% | PQS | Mean f | Min f | Max f | Cov |
| Archaea | 81 | 33,356 | 48.92 | 3137 | 1.76 | 0 | 4.80 | 4.32 |
| Bacteria | 2087 | 49,639 | 48.10 | 112,664 | 0.89 | 0 | 11.51 | 2.11 |
| Eukaryota | 966 | 7951.5 | 43.09 | 104,768 | 1.52 | 0 | 11.44 | 3.93 |
| Group | Seq | Median | GC% | PQS | Mean f | Min f | Max f | Cov |
| Crenarchaeota | 54 | 32,047.5 | 40.91 | 1012 | 1.85 | 0 | 4.80 | 4.76 |
| Euryarchaeota | 27 | 49,107 | 54.92 | 2125 | 1.69 | 0.28 | 3.75 | 3.99 |
| Actinobacteria | 524 | 53,403.5 | 60.90 | 61,313 | 2.27 | 0.33 | 7.02 | 5.12 |
| Bacteroidetes | 32 | 47,060 | 38.12 | 477 | 0.41 | 0.03 | 1.14 | 1.01 |
| Cyanobacteria | 89 | 174,079 | 43.33 | 3875 | 0.82 | 0.06 | 3.88 | 2.10 |
| Deinococcus | 6 | 61,150 | 50.26 | 726 | 4.21 | 0.33 | 11.51 | 10.45 |
| Firmicutes | 527 | 41,843 | 38.14 | 7886 | 0.32 | 0 | 1.39 | 0.78 |
| Proteobacteria | 904 | 49,035 | 50.07 | 38,334 | 0.80 | 0 | 4.55 | 1.90 |
| Amoebozoa | 22 | 495,022 | 42.47 | 21,931 | 0.66 | 0 | 1.89 | 1.60 |
| Arthropoda | 345 | 7276 | 38.77 | 4957 | 0.30 | 0 | 1.92 | 0.73 |
| Chordata | 561 | 7852 | 45.48 | 72,420 | 2.18 | 0 | 11.44 | 5.65 |
| Viridiplantae | 21 | 193,301 | 46.91 | 3542 | 1.06 | 0 | 2.01 | 2.54 |
| Subgroup | Seq | Median | GC% | PQS | Mean f | Min f | Max f | Cov |
| Humans | 120 | 7344 | 42.55 | 15,996 | 1.75 | 0 | 11.44 | 4.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohálová, N.; Cantara, A.; Bartas, M.; Kaura, P.; Šťastný, J.; Pečinka, P.; Fojta, M.; Brázda, V. Tracing dsDNA Virus–Host Coevolution through Correlation of Their G-Quadruplex-Forming Sequences. Int. J. Mol. Sci. 2021, 22, 3433. https://doi.org/10.3390/ijms22073433
Bohálová N, Cantara A, Bartas M, Kaura P, Šťastný J, Pečinka P, Fojta M, Brázda V. Tracing dsDNA Virus–Host Coevolution through Correlation of Their G-Quadruplex-Forming Sequences. International Journal of Molecular Sciences. 2021; 22(7):3433. https://doi.org/10.3390/ijms22073433
Chicago/Turabian StyleBohálová, Natália, Alessio Cantara, Martin Bartas, Patrik Kaura, Jiří Šťastný, Petr Pečinka, Miroslav Fojta, and Václav Brázda. 2021. "Tracing dsDNA Virus–Host Coevolution through Correlation of Their G-Quadruplex-Forming Sequences" International Journal of Molecular Sciences 22, no. 7: 3433. https://doi.org/10.3390/ijms22073433
APA StyleBohálová, N., Cantara, A., Bartas, M., Kaura, P., Šťastný, J., Pečinka, P., Fojta, M., & Brázda, V. (2021). Tracing dsDNA Virus–Host Coevolution through Correlation of Their G-Quadruplex-Forming Sequences. International Journal of Molecular Sciences, 22(7), 3433. https://doi.org/10.3390/ijms22073433