
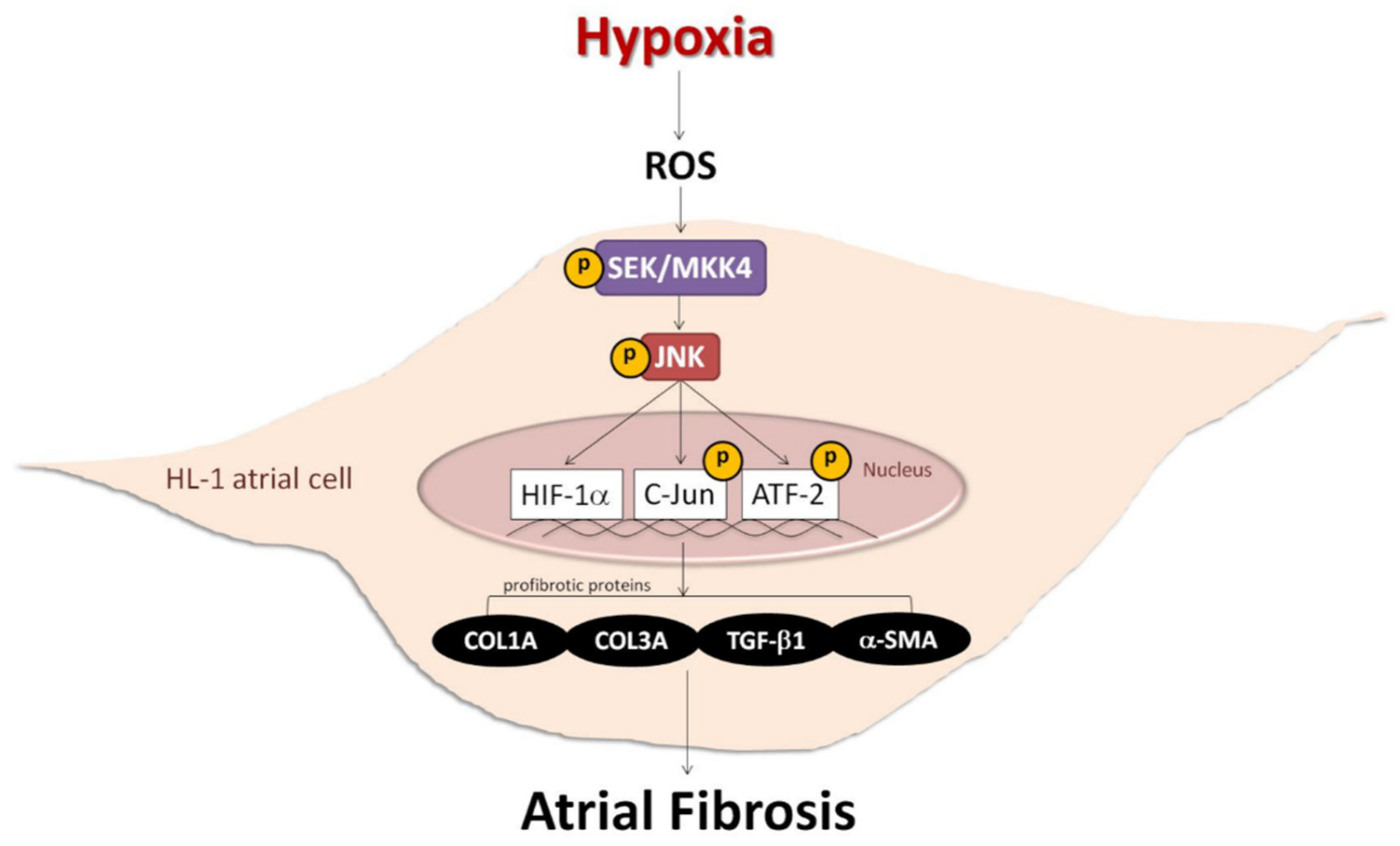
Role of the ROS-JNK Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in HL-1 Cardiomyocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
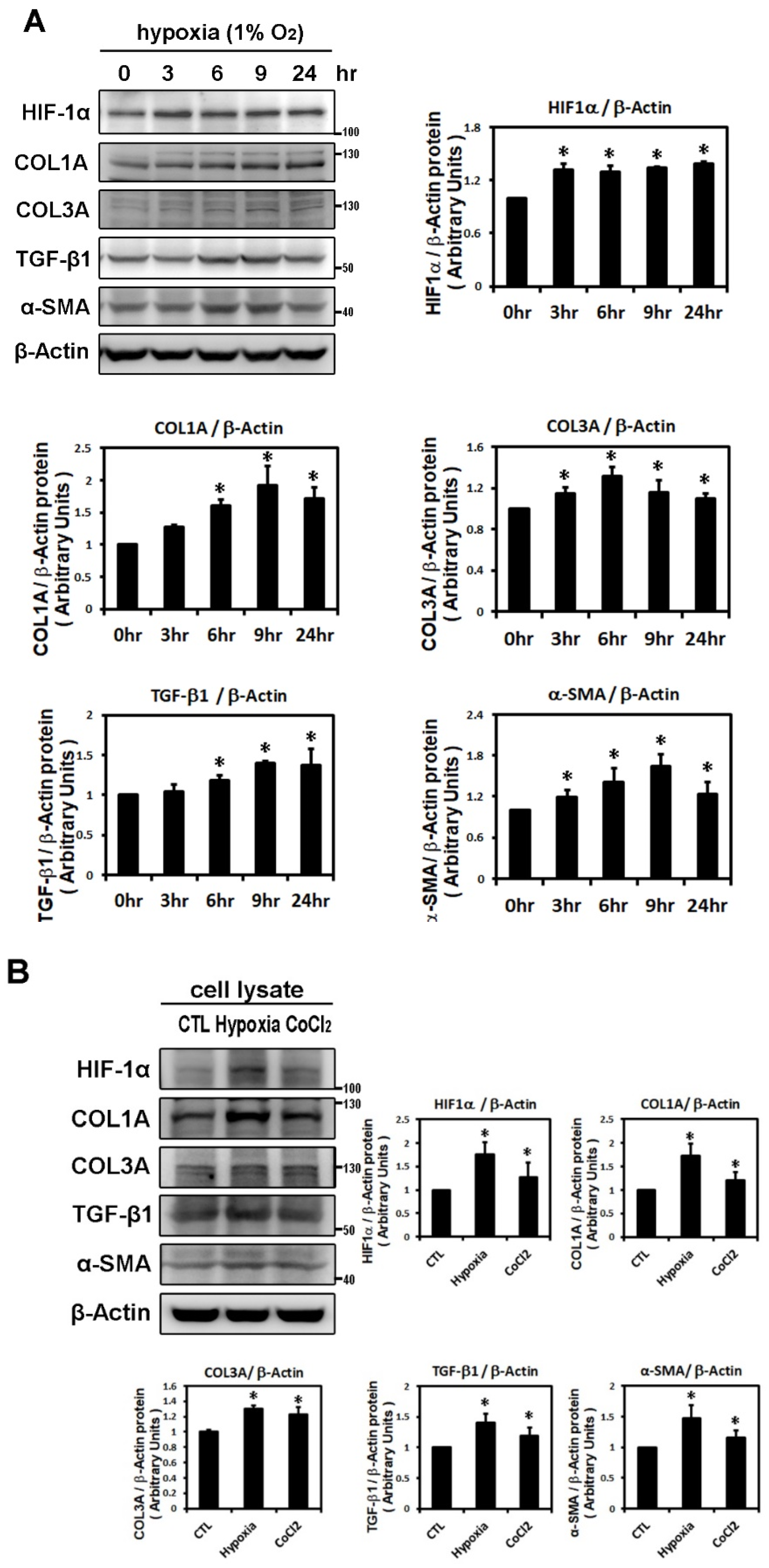
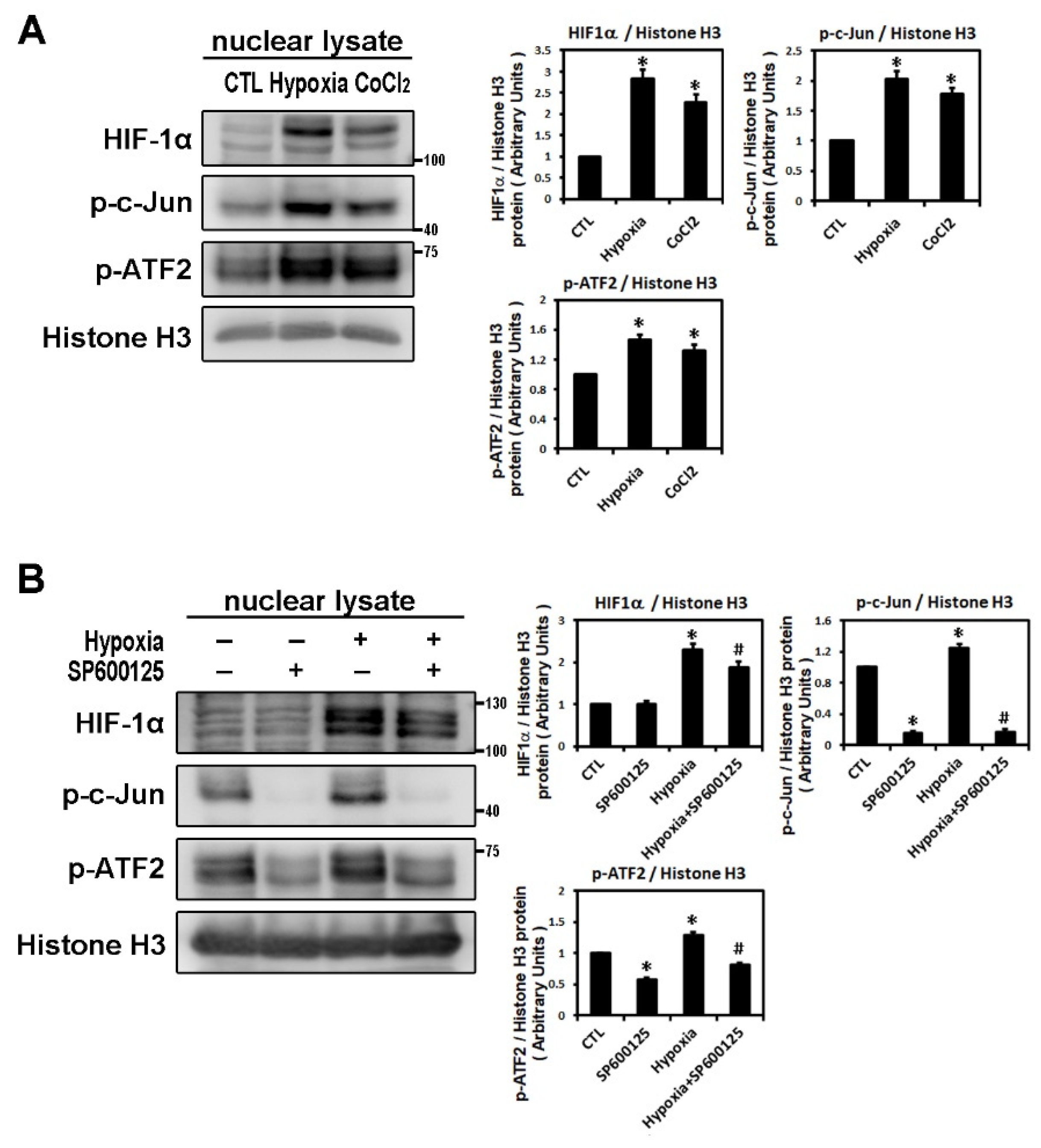
2.1. Effect of Hypoxia on the Expressions of HIF-1α and Profibrotic Proteins
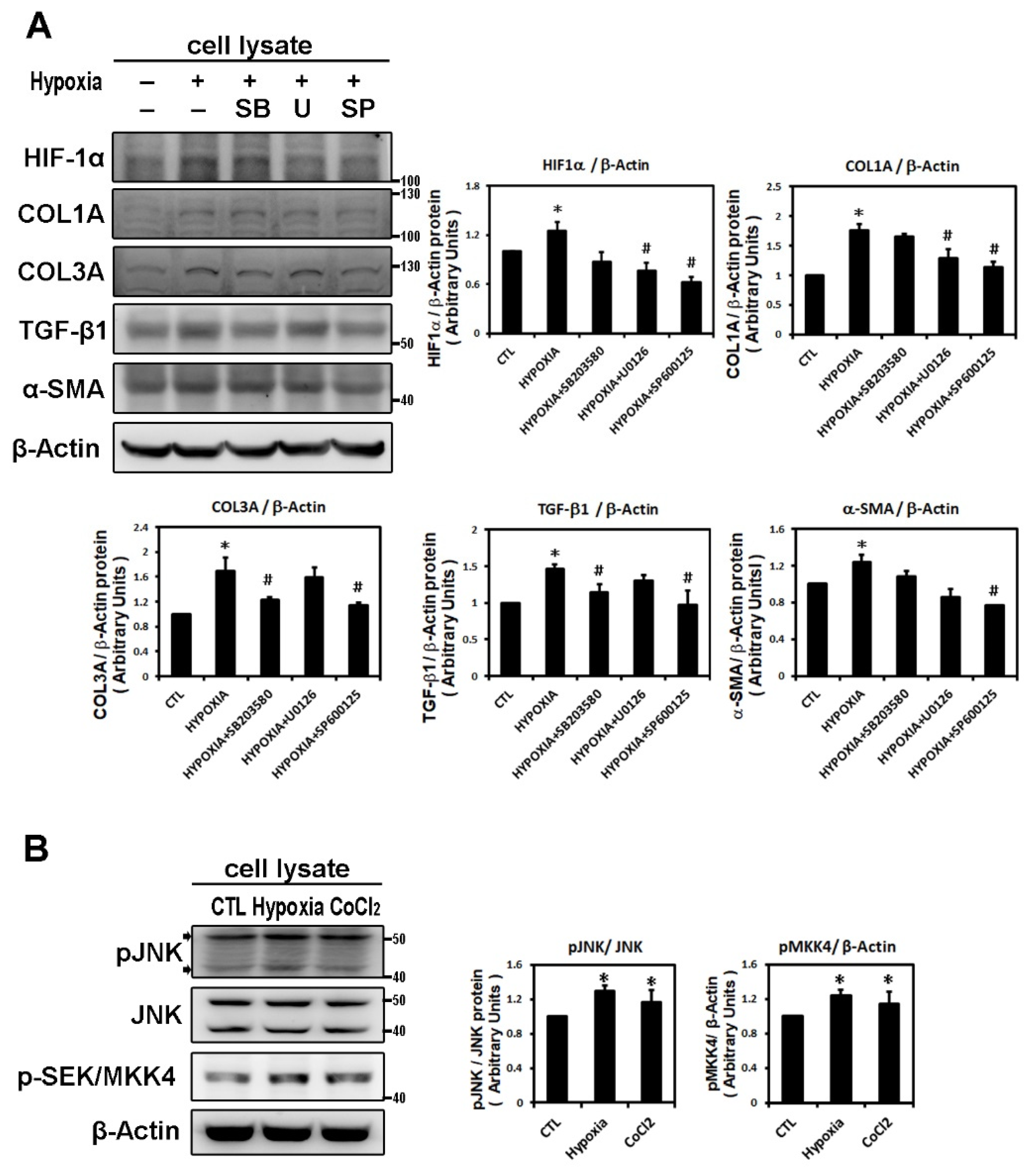
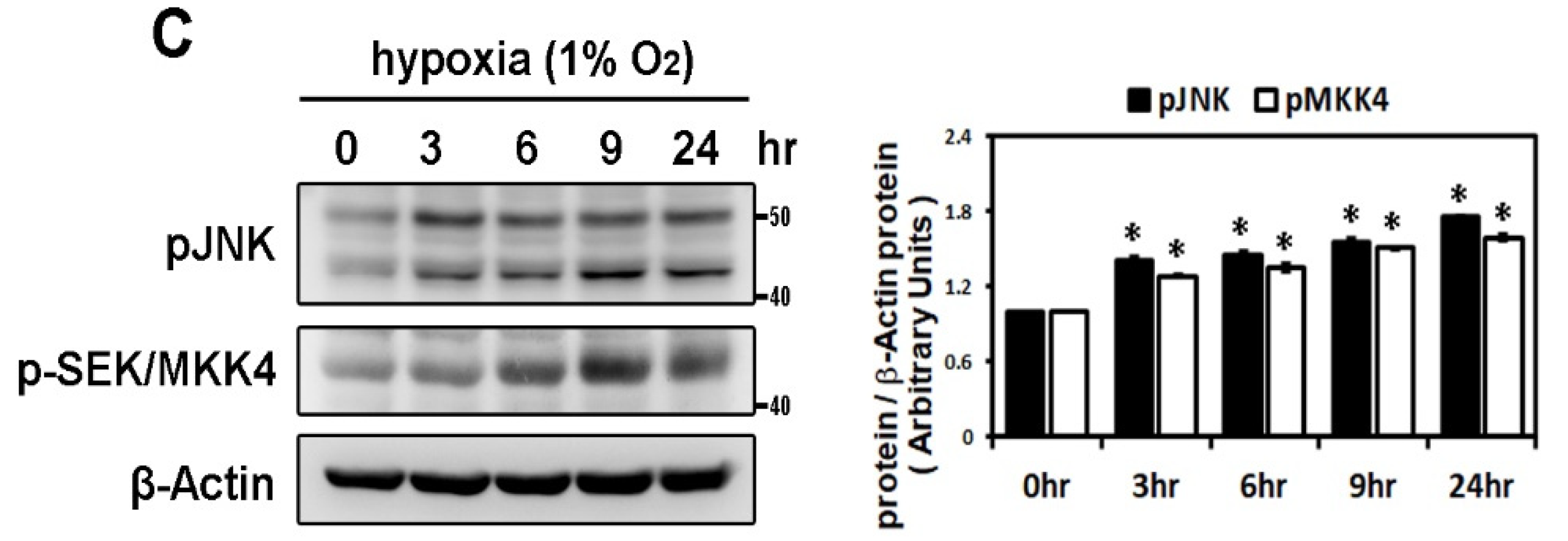
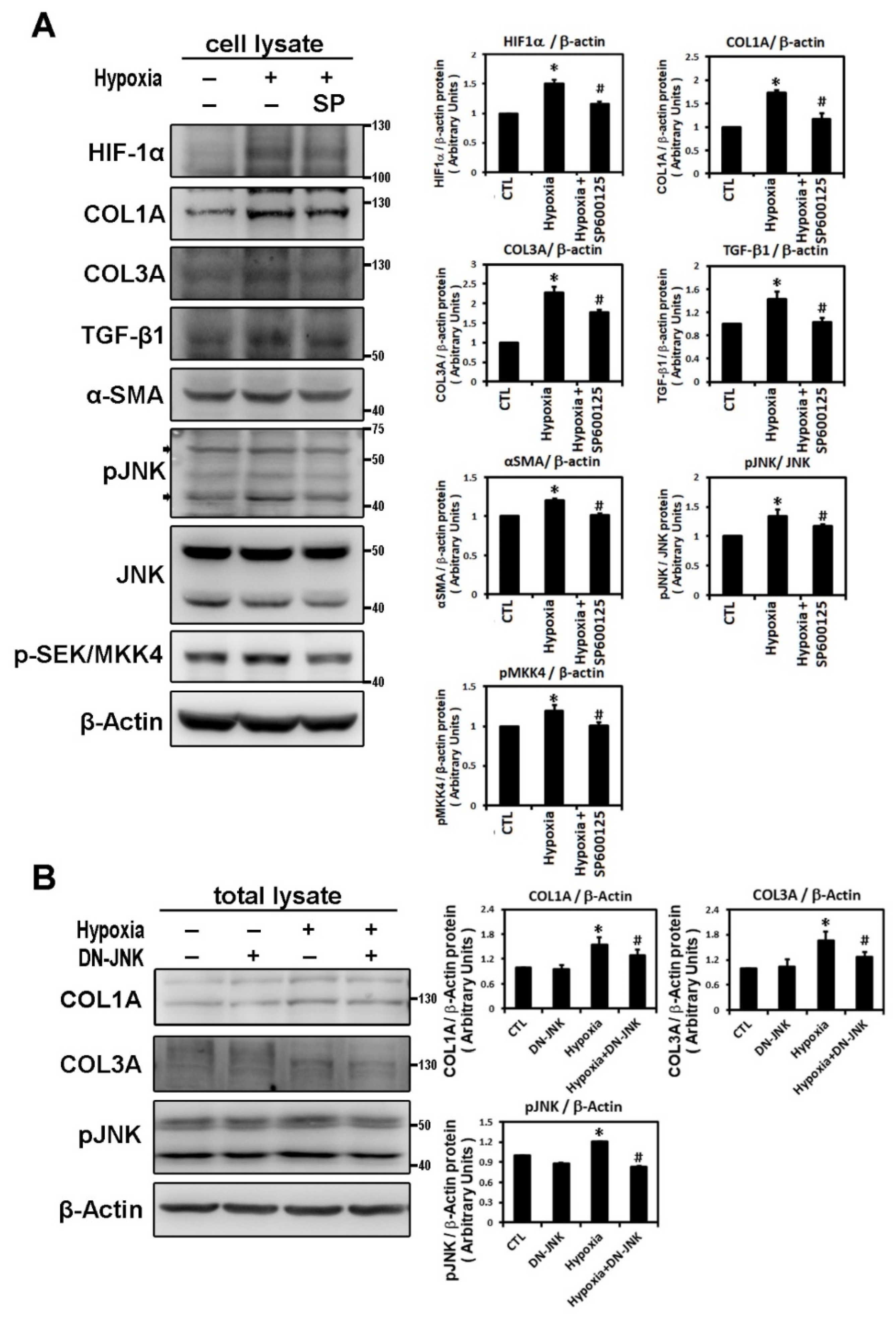
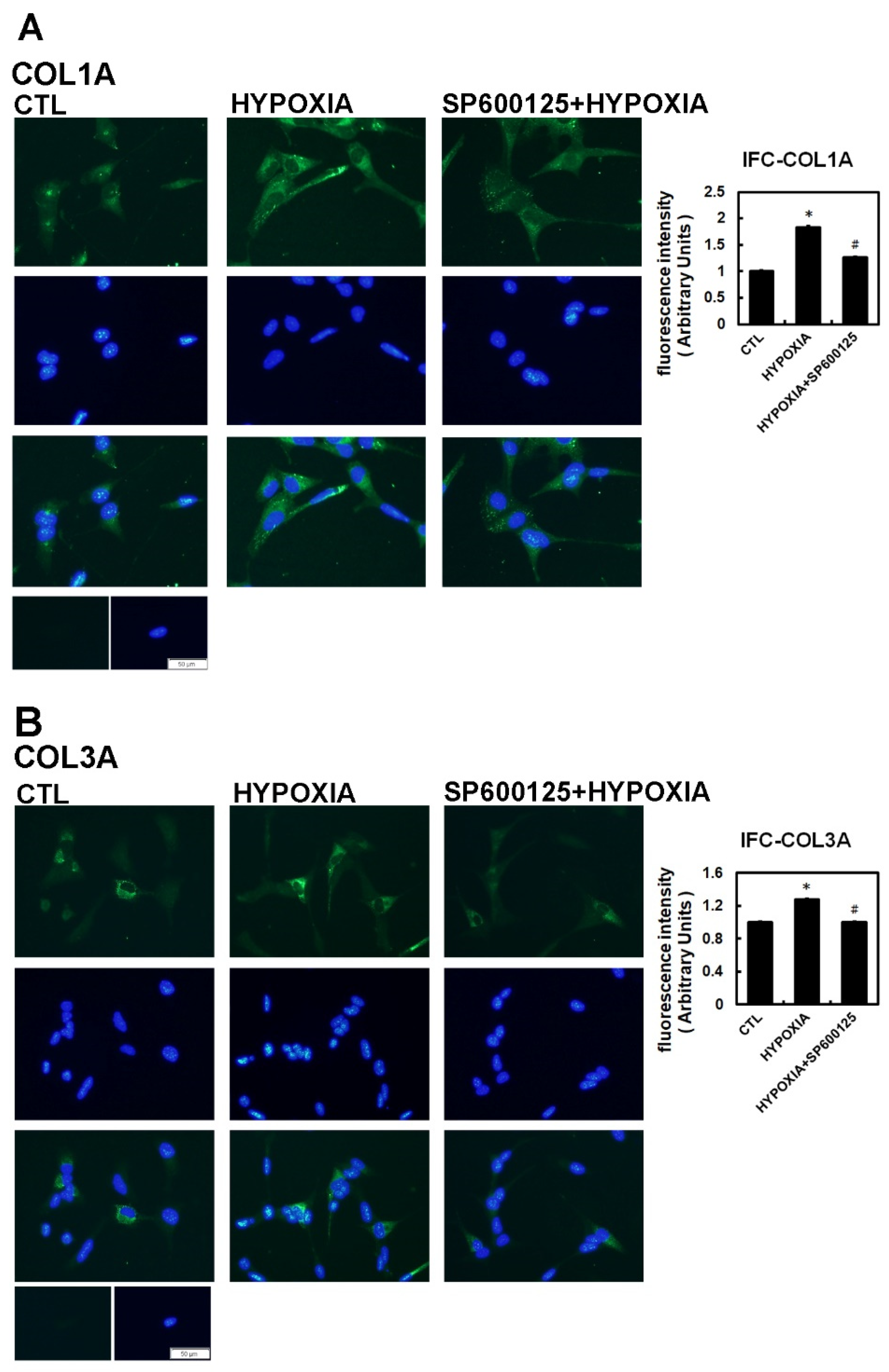
2.2. Hypoxia-Induced Atrial Fibrotic Response via JNK-Dependent Pathways
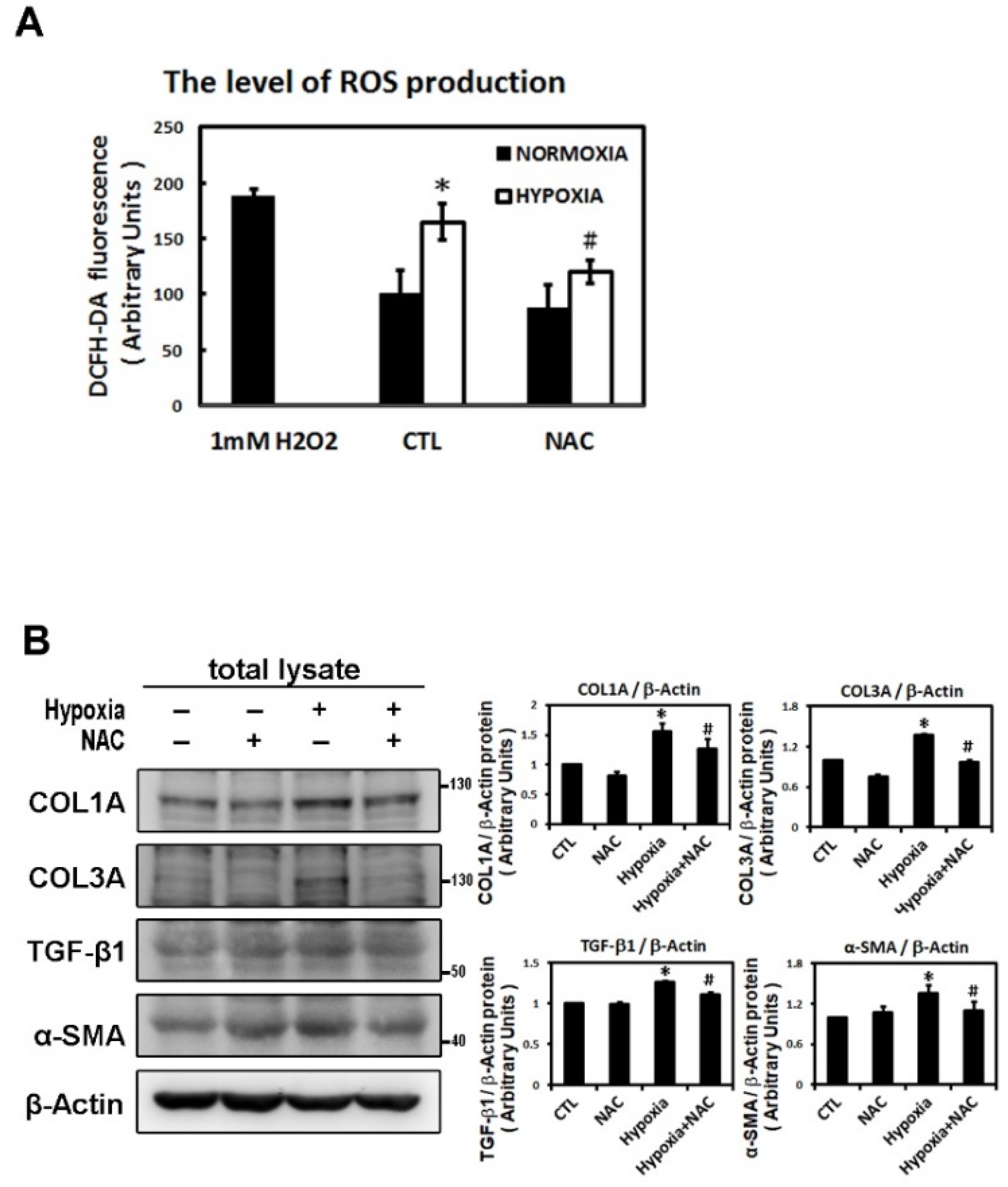
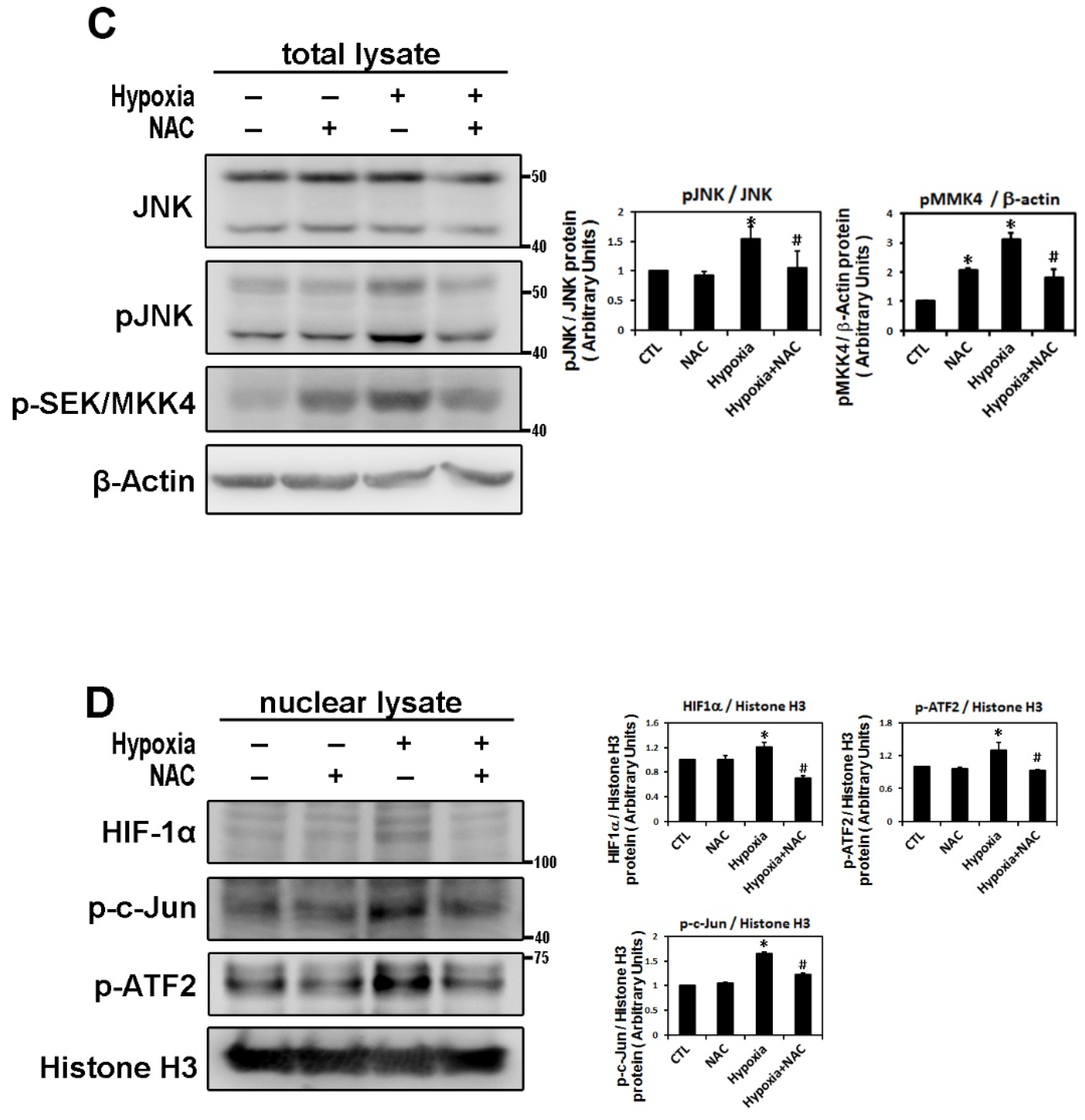
2.3. ROS Modulate Hypoxia-Induced Atrial Profibrotic Proteins Expression
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Hypoxia Stimulation in HL-1 Atrial Myocytes and Drug Preparation
4.2. Transient Transfection
4.3. Protein Extracts and Western Blot Analysis
4.4. Measurement of Reactive Oxygen Species (ROS)
4.5. Immunohistochemical Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Burstein, B.; Nattel, S. Atrial Fibrosis: Mechanisms and Clinical Relevance in Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.P.; Dandapat, A.; Liu, Y.; Hermonat, P.L.; Mehta, J.L. Blockade of hypoxia-reoxygenation-mediated collagen type I expression and MMP activity by overexpression of TGF-beta1 delivered by AAV in mouse cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1833–H1838. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Qi, X.Y.; Michael, G.; Talajic, M.; Nattel, S.; Wakili, R.; Comtois, P.; Chartier, D.; Harada, M.; Iwasaki, Y.-K.; et al. Mechanisms of Atrial Tachyarrhythmias Associated With Coronary Artery Occlusion in a Chronic Canine Model. Circulation 2011, 123, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Tamamori, M.; Ito, H.; Hiroe, M.; Marumo, F.; Hata, R. Stimulation of collagen synthesis in rat cardiac fibroblasts by exposure to hypoxic culture conditions and suppression of the effect by natriuretic peptides. Cell Biol. Int. 1997, 21, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. O2-regulated gene expression: Transcriptional control of cardiorespiratory physiology by HIF-1. J. Appl. Physiol. 2004, 96, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.J.L.; Van Der Velden, H.M.W.; Van Ankeren, E.P.; Ausma, J.; A Allessie, M.; Borgers, M.; Van Eys, G.J.J.M.; Jongsma, H.J. Analysis of altered gene expression during sustained atrial fibrillation in the goat. Cardiovasc. Res. 2002, 54, 427–437. [Google Scholar] [CrossRef]
- Su, F.; Zhang, W.; Chen, Y.; Ma, L.; Zhang, H.; Wang, F. Significance of hypoxia-inducible factor-1α expression with atrial fibrosis in rats induced with isoproterenol. Exp. Ther. Med. 2014, 8, 1677–1682. [Google Scholar] [CrossRef]
- Seta, K.A.; Millhorn, D.E. Functional genomics approach to hypoxia signaling. J. Appl. Physiol. 2004, 96, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, T.; Chen, Y.; Ahokas, R.A.; Sun, Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell. Biochem. 2008, 317, 43–50. [Google Scholar] [CrossRef]
- Erlich, J.R.; Hohnloser, S.H.; Nattel, S. Role of angiotensin system and effects of its inhibition in atrial fibrillation: Clinical and experimental evidence. Eur. Heart J. 2006, 27, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-F.; Yang, S.-F.; Chu, H.-J.; Ueng, K.-C. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int. J. Cardiol. 2013, 169, 17–28. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Tanaka, K.; Honda, M.; Takabatake, T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J. Am. Coll. Cardiol. 2001, 37, 676–685. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Gramley, F.; Lorenzen, J.; Jedamzik, B.; Gatter, K.; Koellensperger, E.; Munzel, T.; Pezzella, F. Atrial fibrillation is associated with cardiac hypoxia. Cardiovasc. Pathol. 2010, 19, 102–111. [Google Scholar] [CrossRef]
- Basu, R.K.; Hubchak, S.; Hayashida, T.; Runyan, C.E.; Schumacker, P.T.; Schnaper, H.W. Interdependence of HIF-1α and TGF-β/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am. J. Physiol. Physiol. 2011, 300, F898–F905. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L.; Kaska, S.; Wentling, C. Hypoxia-Inducible Factor Activation in Myeloid Cells Contributes to the Development of Liver Fibrosis in Cholestatic Mice. J. Pharmacol. Exp. Ther. 2012, 341, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced liver fibrosis in hypoxia-inducible factor-1α-deficient mice. Am. J. Physiol. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Liu, Y. Targeting Hypoxia Inducible Factors-1α As a Novel Therapy in Fibrosis. Front. Pharmacol. 2017, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Sugden, P.H.; Clerk, A. Stress-Responsive Mitogen-Activated Protein Kinases (c-Jun N-Terminal Kinases and p38 Mitogen-Activated Protein Kinases) in the Myocardium. Circ. Res. 1998, 83, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Kuo, W.W.; Wu, H.C.; Lai, T.Y.; Wu, C.H.; Hwang, J.M.; Wang, W.-H.; Tsai, F.-J.; Yang, J.-J.; Huang, C.-Y.; et al. ZAK induces MMP-2 activity via JNK/p38 signals and reduces MMP-9 activity by increasing TIMP-1/2 expression in H9c2 cardiomyoblast cells. Mol. Cell Biochem. 2009, 325, 69–77. [Google Scholar] [CrossRef]
- Gu, J.; Liu, X.; Wang, Q.X.; Tan, H.W.; Guo, M.; Jiang, W.F.; Zhou, L. Angiotensin II increases CTGF expression via MAPKs/TGF-beta1/TRAF6 pathway in atrial fibroblasts. Exp. Cell Res. 2012, 318, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yi, X.; Ma, L.; Zhou, Y. Hepatocyte growth factor and basic fibroblast growth factor regulate atrial fibrosis in patients with atrial fibrillation and rheumatic heart disease via the mitogen-activated protein kinase signaling pathway. Exp. Ther. Med. 2013, 6, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Reynolds, C.; Jo, S.; Wersto, R.; Han, Q.; Xiao, R. Intracellular acidosis-activated p38 MAPK signaling and its essential role in cardiomyocyte hypoxic injury. FASEB J. 2004, 19, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, H.; Perrino, C.; Takaoka, H.; Davis, R.J.; Prasad, S.V.N.; Rockman, H.A. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem. Biophys. Res. Commun. 2006, 343, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Kyoi, S.; Otani, H.; Matsuhisa, S.; Akita, Y.; Tatsumi, K.; Enoki, C.; Fujiwara, H.; Imamura, H.; Kamihata, H.; Iwasaka, T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 2006, 69, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kim, S.; Izumi, Y.; Yoshida, K.; Yoshiyama, M.; Matsuzawa, A.; Ichijo, H.; Iwao, H. Apoptosis Signal-Regulating Kinase 1 Plays a Pivotal Role in Angiotensin II–Induced Cardiac Hypertrophy and Remodeling. Circ. Res. 2003, 93, 874–883. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Subramanian, V.; Singh, M.; Singh, K. β1 Integrins modulate β-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension 2007, 49, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Bergami, P.R.L.; Lau, E.; Ronai, Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK Signaling Pathway in Renal Fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Pant, I.; Rao, S.G.; Kondaiah, P. Role of areca nut induced JNK/ATF2/Jun axis in the activation of TGF-β pathway in precancerous Oral Submucous Fibrosis. Sci. Rep. 2016, 6, srep34314. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.-Q.; Ma, A.; He, J.-Z.; Wang, L.; Jia, Y.-L.; Shao, G.-Y.; Li, M.; Zhou, H.; Lin, S.-Q.; Ran, J.-H.; et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-β/Smad and MAPK signaling pathways. Acta Pharmacol. Sin. 2019, 41, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Reil, J.-C.; Hohl, M.; Oberhofer, M.; Kazakov, A.; Kaestner, L.; Mueller, P.; Adam, O.; Maack, C.; Lipp, P.; Mewis, C.; et al. Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodelling. Cardiovasc. Res. 2010, 87, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Theobald, K.; Lavall, D.; Grube, M.; Kroemer, H.K.; Ameling, S.; Schäfers, H.-J.; Böhm, M.; Laufs, U. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J. Mol. Cell. Cardiol. 2011, 50, 678–685. [Google Scholar] [CrossRef]
- Shih, Y.-C.; Chen, C.-L.; Zhang, Y.; Mellor, R.L.; Kanter, E.M.; Fang, Y.; Wang, H.-C.; Hung, C.-T.; Nong, J.-Y.; Chen, H.-J.; et al. Endoplasmic Reticulum Protein TXNDC5 Augments Myocardial Fibrosis by Facilitating Extracellular Matrix Protein Folding and Redox-Sensitive Cardiac Fibroblast Activation. Circ. Res. 2018, 122, 1052–1068. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Laderoute, K.R.; A Webster, K. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circ. Res. 1997, 80, 336–344. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Jiang, H.; Gao, W.; Wu, J.; Peng, K.; Shi, Y.-F.; Zhang, X.-J. The JNK/AP1/ATF2 pathway is involved in H2O2-induced acetylcholinesterase expression during apoptosis. Cell. Mol. Life Sci. 2008, 65, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G Protein-coupled Receptor 30 Is Up-regulated by Hypoxia-inducible Factor-1α (HIF-1α) in Breast Cancer Cells and Cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Xu, M.; Yuan, Y.; Li, F.F.; Yang, Z.; Liu, Y.; Zhou, M.; Bian, Z.; Deng, W.; Gao, L.; et al. Cathepsin b deficiency attenuates cardiac remodeling in response to pressure overload via tnf-alpha/ask1/jnk pathway. Am. J. Physiol. Heart. Circ. Physiol. 2015, 308, H1143–H1154. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yang, Y.; Xue, C.; Tan, M.; Xu, L.; Gao, J.; Xu, L.; Zong, J.; Qian, W. Zinc finger protein zbtb20 protects against cardiac remodelling post-myocardial infarction via ros-tnfalpha/ask1/jnk pathway regulation. J. Cell Mol. Med. 2020, 24, 13383–13396. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-F.; Yang, S.-F.; Lo, C.-H.; Chu, H.-J.; Ueng, K.-C. Role of the ROS-JNK Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in HL-1 Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 3249. https://doi.org/10.3390/ijms22063249
Tsai C-F, Yang S-F, Lo C-H, Chu H-J, Ueng K-C. Role of the ROS-JNK Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in HL-1 Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(6):3249. https://doi.org/10.3390/ijms22063249
Chicago/Turabian StyleTsai, Chin-Feng, Shun-Fa Yang, Chien-Hsien Lo, Hsiao-Ju Chu, and Kwo-Chang Ueng. 2021. "Role of the ROS-JNK Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in HL-1 Cardiomyocytes" International Journal of Molecular Sciences 22, no. 6: 3249. https://doi.org/10.3390/ijms22063249
APA StyleTsai, C.-F., Yang, S.-F., Lo, C.-H., Chu, H.-J., & Ueng, K.-C. (2021). Role of the ROS-JNK Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in HL-1 Cardiomyocytes. International Journal of Molecular Sciences, 22(6), 3249. https://doi.org/10.3390/ijms22063249