
GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
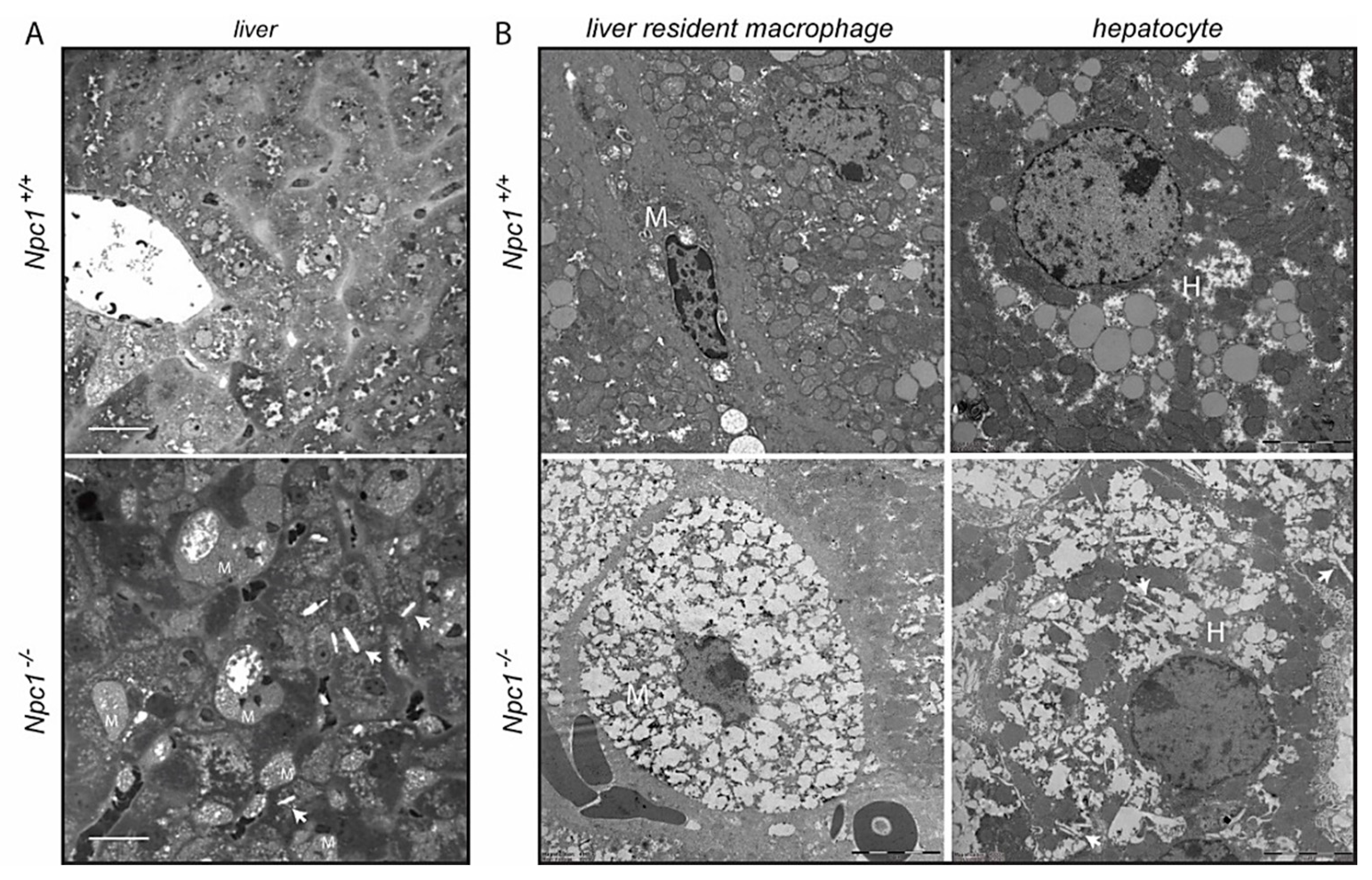
2.1. Structural Analysis of Npc1−/− Liver
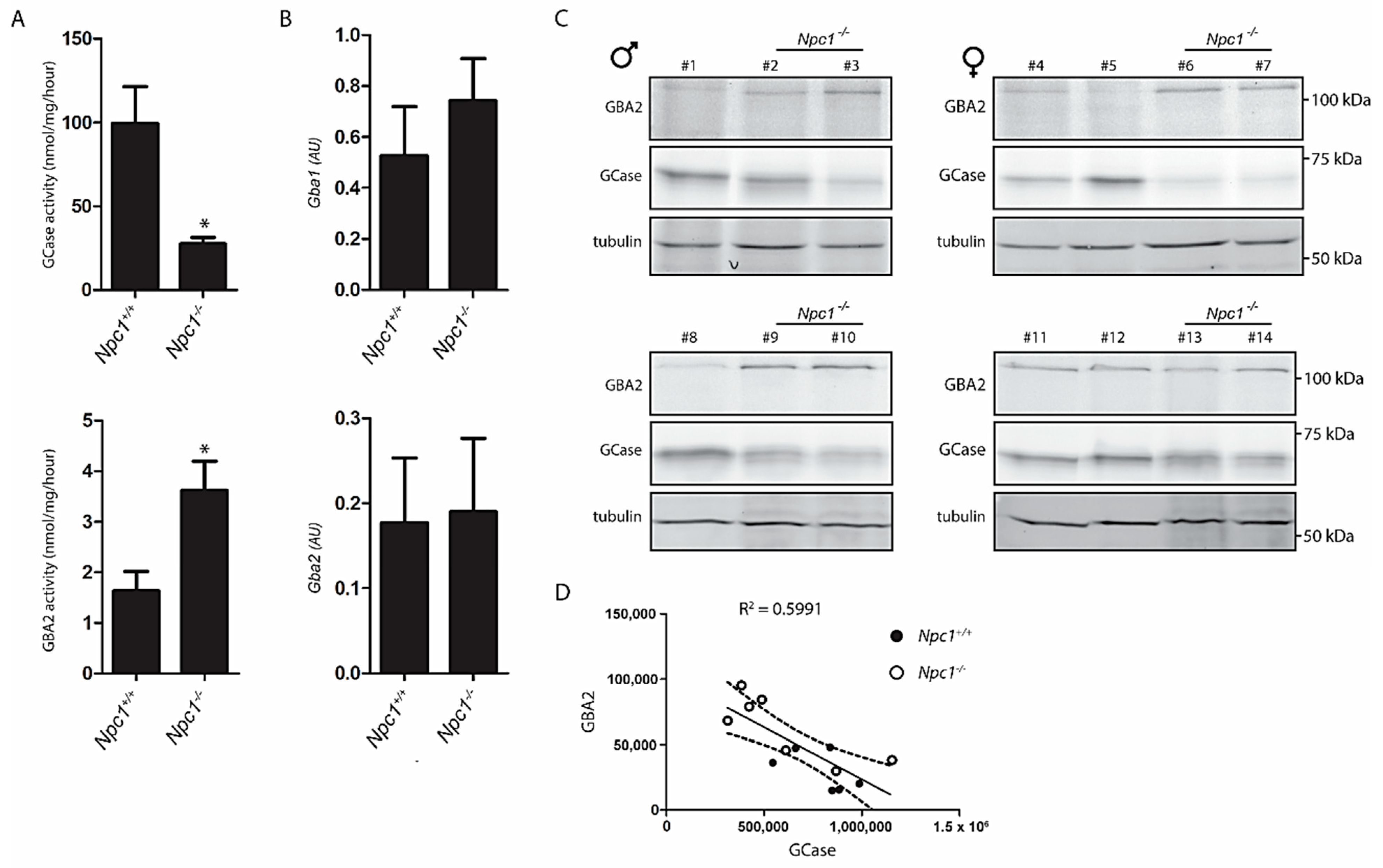
2.2. Lysosomal GCase and Cytosol Facing GBA2 in NPC Liver
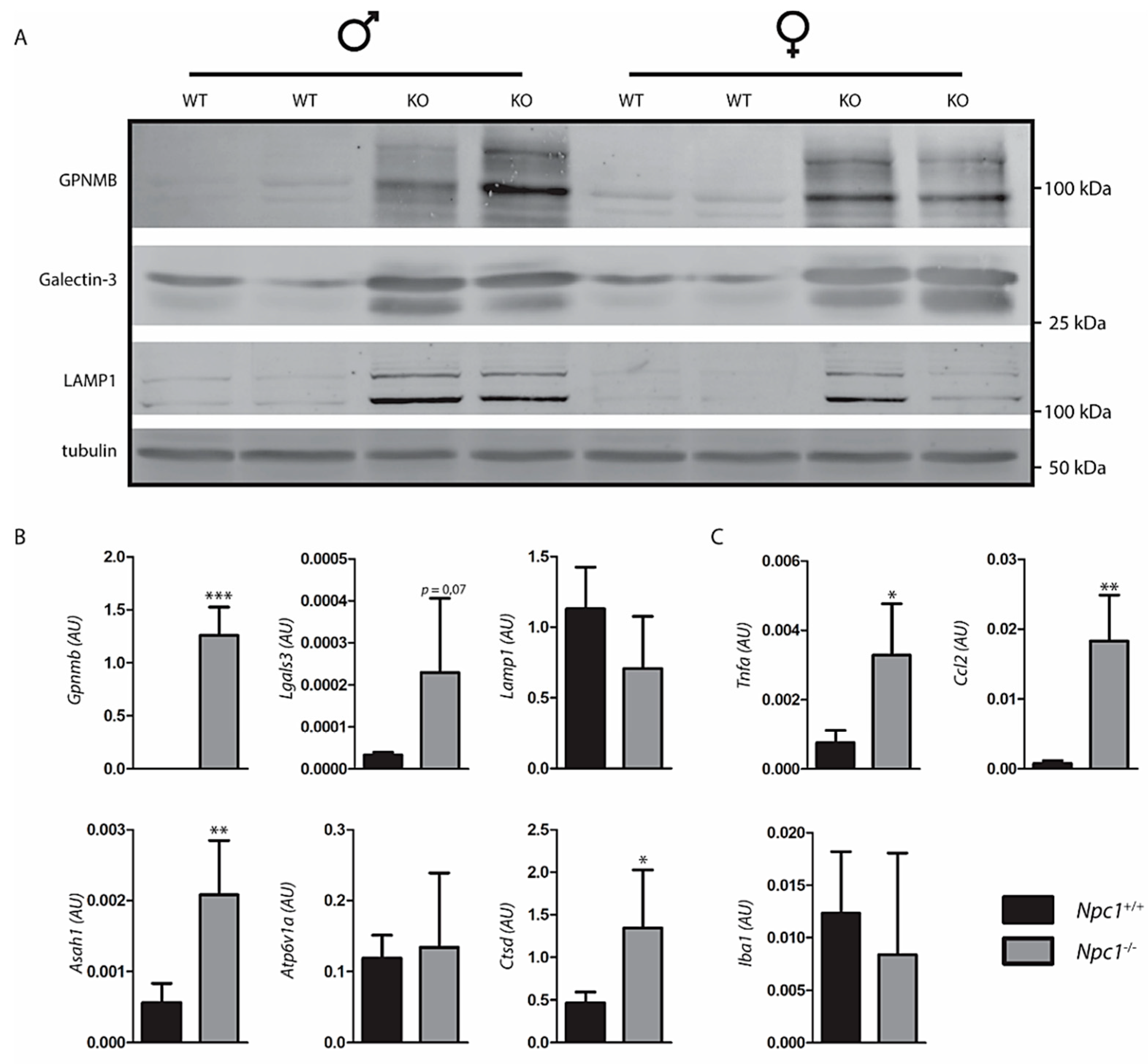
2.3. Response to Lysosomal Storage in NPC Liver
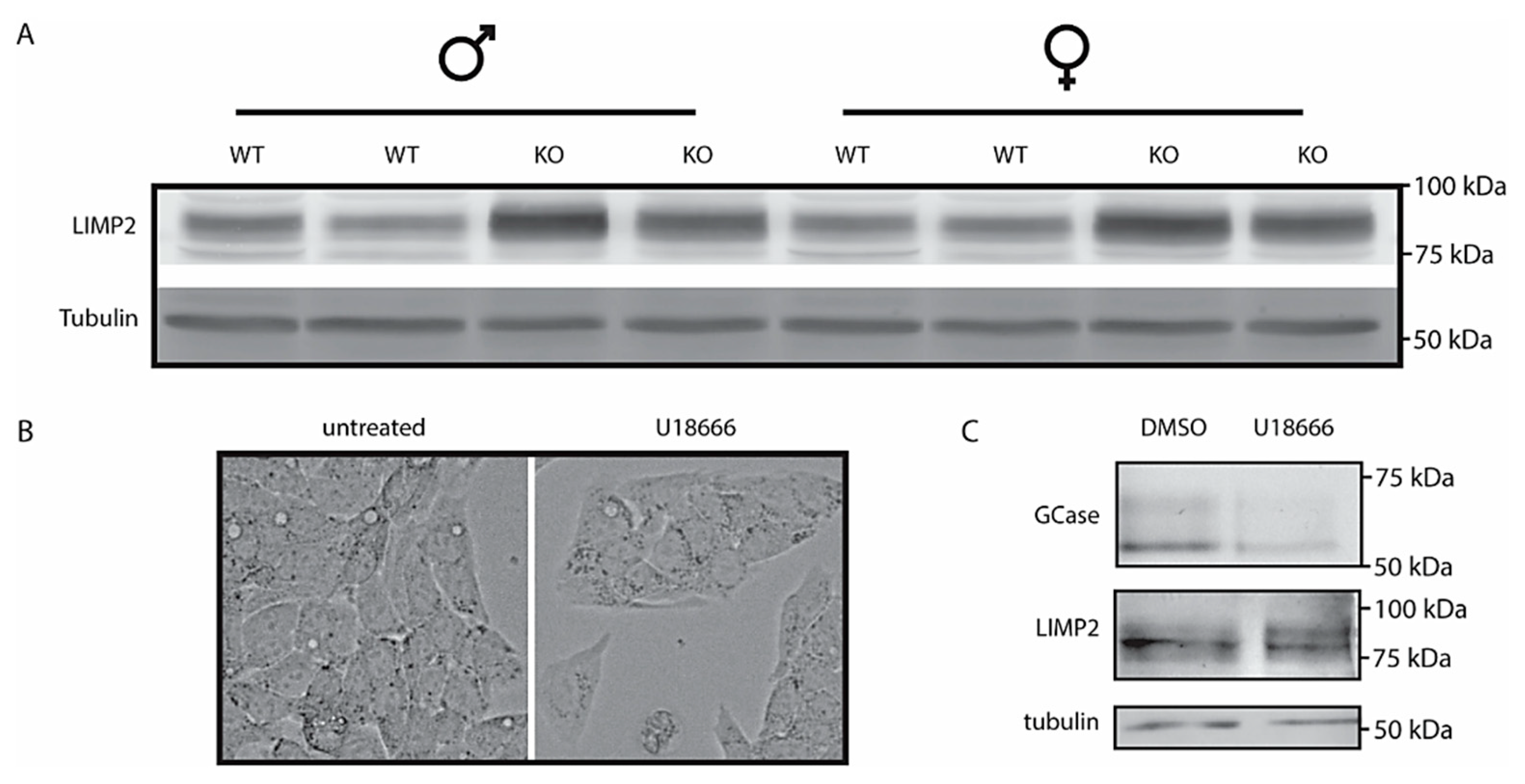
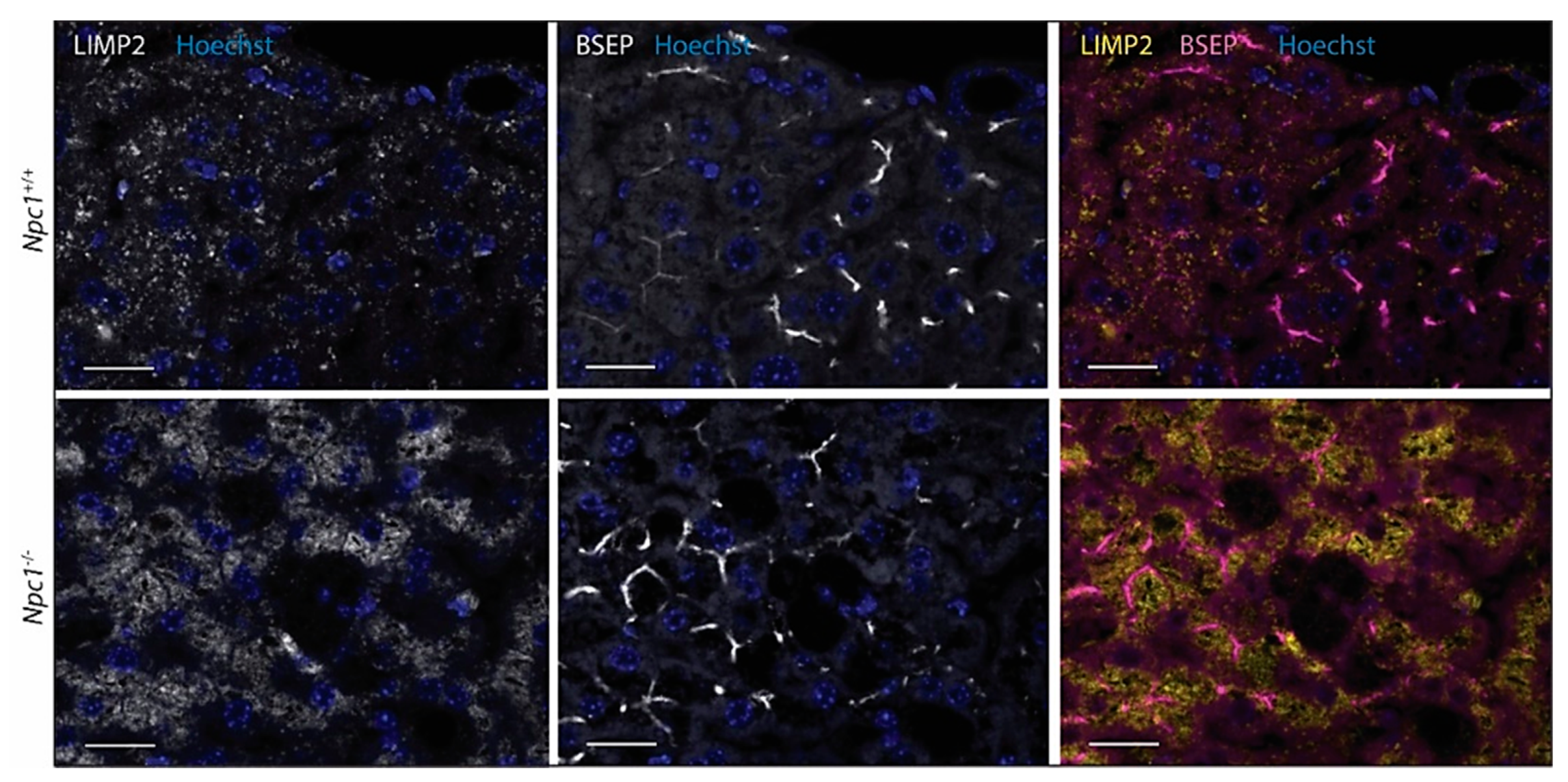
2.4. LIMP2 Upregulation in NPC Liver but Not in Cultured Cells with Pharmacologically Induced Lysosomal Cholesterol Accumulation
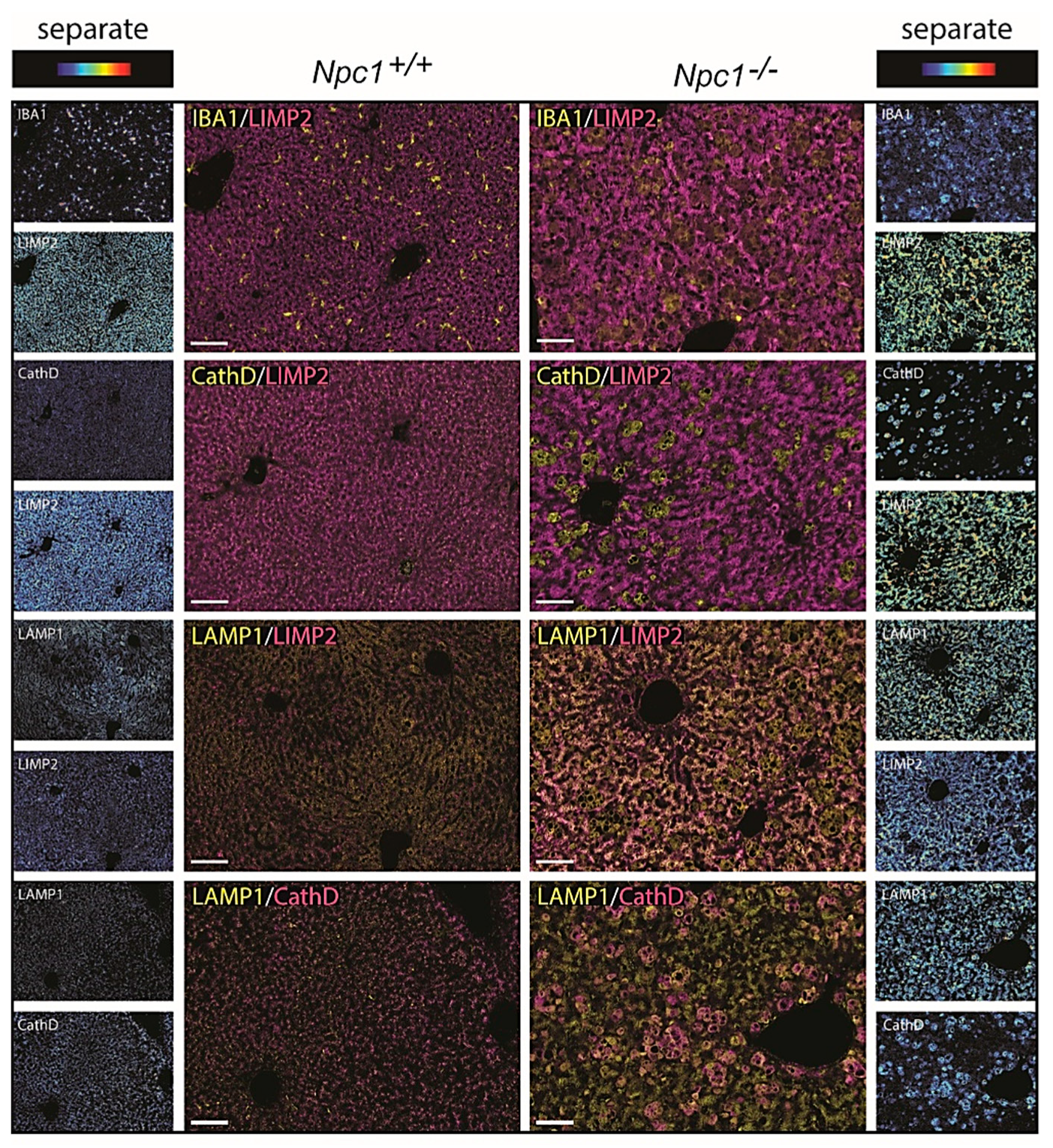
2.5. Immunohistochemical Analysis of NPC Liver
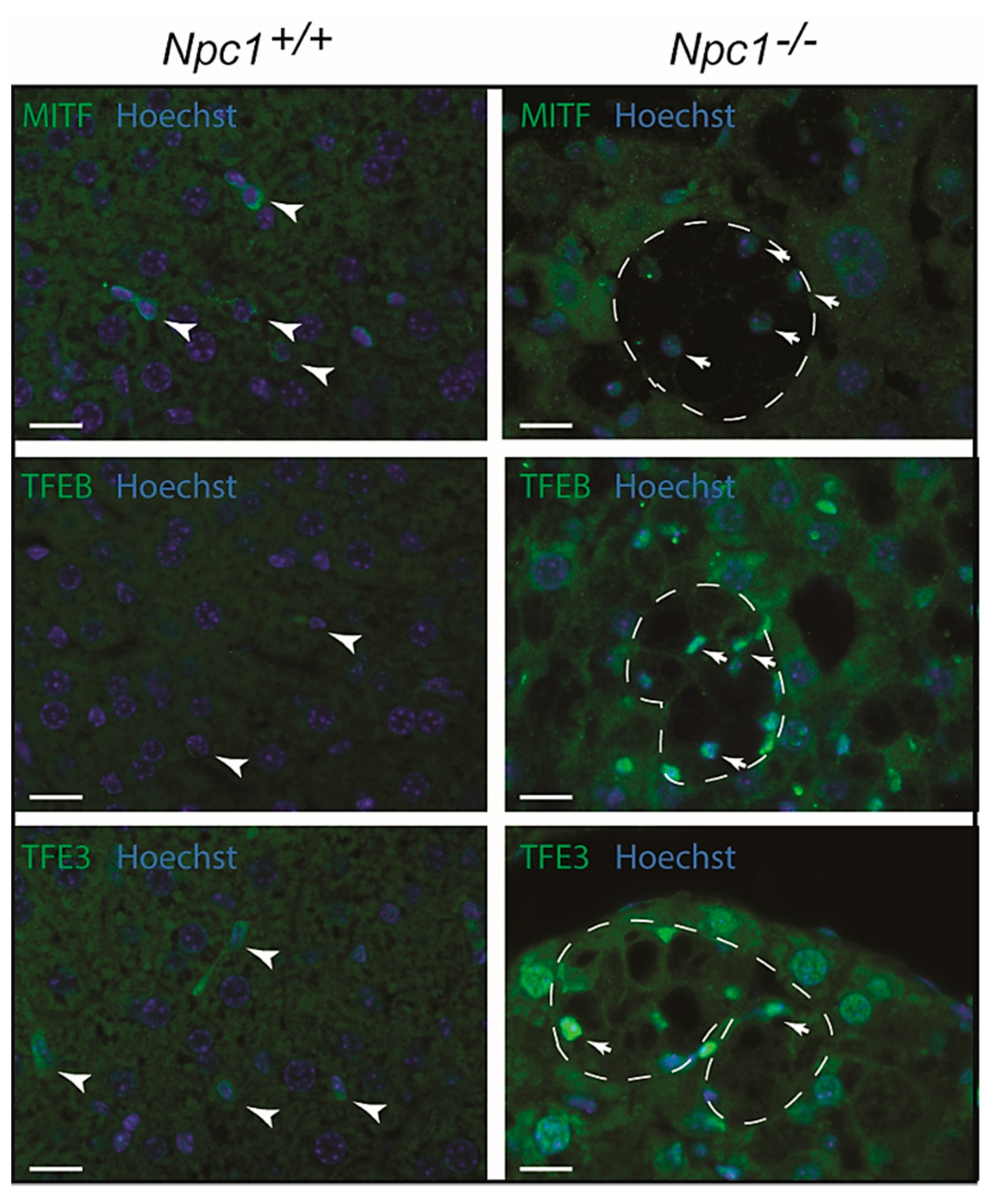
2.6. MiT/TFE Transcription Factors Regulating Lysosomal Biogenesis in NPC Liver
3. Discussion
4. Materials and Methods
4.1. Cell Culture Experiments
4.2. Animals
4.3. Electron Microscopy and Toluidine Blue Staining
4.4. Immunohistochemistry and Immunofluorescence
4.5. Enzyme Activity Assays
4.6. Activity-Based Probe Analysis
4.7. Western Blot Analysis
4.8. RNA Extraction and Real-Time PCR
4.9. Lipid Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABP | activity based probe |
| AMRF | acute myoclonus renal failure syndrome |
| CCL2 | C−C motif chemokine ligand 2 |
| CCL18 | C−C motif chemokine ligand 18 |
| GBA2 | glucosylceramidase beta 2 |
| GCase | glucosylceramide beta |
| GD | Gaucher disease |
| GlcCer | glucosylceramide |
| GlcChol | glucosylated cholesterol |
| GlcSph | glucosylsphingosine |
| GPNMB | glycoprotein non metastatic protein B |
| HMG-CoA reductase | 3-hydroxy-3-methylglutaryl-CoA reductase |
| IBA1 | allograft inflammatory factor 1 |
| LAMP1 | lysosomal associated membrane protein 1 |
| LIMP2 | sterol regulatory element binding transcription factor 2 |
| MiT/TFE | microphthalmia-transcription factor E |
| MITF | melanocyte inducing transcription factor |
| NPC | Niemann–Pick type C |
| NPC1 | NPC intracellular cholesterol transporter 1 |
| NPC2 | NPC intracellular cholesterol transporter 2 |
| SREBP2 | scavenger receptor class B member 2 |
| TFE3 | transcription factor E3 |
| TEM | transmission electron microscopy |
| TFEB | transcription factor EB |
| TNFα | tumor necrosis factor alpha |
| ZKSCAN3 | zinc finger with KRAB and SCAN domains 3 |
References
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Elleder, M.; Šmíd, F.; Hyniová, H.; Čihula, J.; Zeman, J.; Macek, M. Liver findings in Niemann-Pick disease type C. Histochem. J. 1984, 16, 1147–1170. [Google Scholar] [CrossRef] [PubMed]
- Pentchev, P.G.; Gal, A.E.; Booth, A.D.; Omodeo-Sale, F.; Fours, J.; Neumeyer, B.A.; Quirk, J.M.; Dawson, G.; Brady, R.O. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim. Biophys. Acta Lipids Lipid Metab. 1980, 619, 669–679. [Google Scholar] [CrossRef]
- Vanier, M.T. Biochemical studies in niemann-pick disease I. Major sphingolipids of liver and spleen. Biochim. Biophys. Acta Lipids Lipid Metab. 1983, 750, 178–184. [Google Scholar] [CrossRef]
- Salvioli, R.; Scarpa, S.; Ciaffoni, F.; Tatti, M.; Ramoni, C.; Vanier, M.T.; Vaccaro, A.M. Glucosylceramidase Mass and Subcellular Localization Are Modulated by Cholesterol in Niemann-Pick Disease Type C. J. Biol. Chem. 2004, 279, 17674–17680. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.; Gaspar, P.; Mirzaian, M.; van Roomen, C.; Ottenhoff, R.; Alfonso, P.; Irún, P.; Giraldo, P.; Wisse, P.; et al. Lyso-glycosphingolipid abnormalities in different murine models of lysosomal storage disorders. Mol. Genet. Metab. 2016, 117, 186–193. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van Der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular β-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Aten, J.; Ottenhoff, R.; Van Roomen, C.P.A.A.; Moro, D.H.; Claessen, N.; Veloz, M.F.V.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 Activity Ameliorates Neuropathology in Niemann-Pick Type C Mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Zachos, C.; Blanz, J.; Saftig, P.; Schwake, M. A Critical Histidine Residue Within LIMP-2 Mediates pH Sensitive Binding to Its Ligand β-Glucocerebrosidase. Traffic 2012, 13, 1113–1123. [Google Scholar] [CrossRef]
- Schwake, M.; Schröder, B.; Saftig, P. Lysosomal Membrane Proteins and Their Central Role in Physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.-J.; Weng, X.; Kim, Y.A.; Collins, R.; Trimble, W.; et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pergande, M.R.; Zarate, E.; Haney-Ball, C.; Davidson, C.D.; Scesa, G.; I Givogri, M.; Bongarzone, E.R.; Cologna, S.M. Standard-flow LC and thermal focusing ESI elucidates altered liver proteins in late stage Niemann–Pick, type C1 disease. Bioanalysis 2019, 11, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Pergande, M.R.; Nguyen, T.T.A.; Haney-Ball, C.; Davidson, C.D.; Cologna, S.M. Quantitative, Label-Free Proteomics in the Symptomatic Niemann–Pick, Type C1 Mouse Model Using Standard Flow Liquid Chromatography and Thermal Focusing Electrospray Ionization. Proteomics 2019, 19, e1800432. [Google Scholar] [CrossRef]
- Hollak, C.E.; Van Weely, S.; Van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; De Fost, M.; Hollak, C.E.M.; Maas, M.; Bleijlevens, B.; Van Breemen, M.J.; Van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Moro, D.H.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.; Aerts, J. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 811–825. [Google Scholar] [CrossRef]
- Kramer, G.; Wegdam, W.; Donker-Koopman, W.; Ottenhoff, R.; Gaspar, P.; Verhoek, M.; Nelson, J.; Gabriel, T.; Kallemeijn, W.; Boot, R.G.; et al. Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio 2016, 6, 902–913. [Google Scholar] [CrossRef]
- Van Der Lienden, M.J.; Gaspar, P.; Boot, R.; Aerts, J.M.; Van Eijk, M. Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages. Int. J. Mol. Sci. 2018, 20, 66. [Google Scholar] [CrossRef]
- Aerts, J.M.; Kuo, C.-L.; Lelieveld, L.T.; Boer, D.E.; Van Der Lienden, M.J.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Guo, Y.; He, W.; Boer, A.M.; Wevers, R.; De Bruijn, A.; Groener, J.; Hollak, C.; Aerts, J.; Galjaard, H.; Van Diggelen, O. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J. Inherit. Metab. Dis. 1995, 18, 717–722. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Gabriel, T.L.; Aten, J.; Van Roomen, C.P.A.A.; Ottenhoff, R.; Claessen, N.; Alfonso, P.; Irún, P.; Giraldo, P.; Aerts, J.M.F.G.; et al. Gpnmb Is a Potential Marker for the Visceral Pathology in Niemann-Pick Type C Disease. PLoS ONE 2016, 11, e0147208. [Google Scholar] [CrossRef]
- Overkleeft, H.S.; Renkema, G.H.; Neele, J.; Vianello, P.; Hung, I.O.; Strijland, A.; Van Der Burg, A.M.; Koomen, G.-J.; Pandit, U.K.; Aerts, J.M.F.G. Generation of Specific Deoxynojirimycin-type Inhibitors of the Non-lysosomal Glucosylceramidase. J. Biol. Chem. 1998, 273, 26522–26527. [Google Scholar] [CrossRef] [PubMed]
- Abela, G.S.; Aziz, K.; Vedre, A.; Pathak, D.R.; Talbott, J.D.; DeJong, J. Effect of Cholesterol Crystals on Plaques and Intima in Arteries of Patients with Acute Coronary and Cerebrovascular Syndromes. Am. J. Cardiol. 2009, 103, 959–968. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Landis, C.S.; Jin, G.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated with Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Bocan, T.M.; Schifani, T.A.; Guyton, J.R. Ultrastructure of the human aortic fibrolipid lesion. Formation of the atherosclerotic lipid-rich core. Am. J. Pathol. 1986, 123, 413–424. [Google Scholar]
- Gabriel, T.L.; Tol, M.J.; Ottenhof, R.; Van Roomen, C.; Aten, J.; Claessen, N.; Hooibrink, B.; De Weijer, B.; Serlie, M.J.; Argmann, C.; et al. Lysosomal Stress in Obese Adipose Tissue Macrophages Contributes to MITF-Dependent Gpnmb Induction. Diabetes 2014, 63, 3310–3323. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Sakai, J.; A Duncan, E.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-Regulated Release of SREBP-2 from Cell Membranes Requires Two Sequential Cleavages, One Within a Transmembrane Segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Gaspar, P.; Kallemeijn, W.W.; Strijland, A.; Scheij, S.; Van Eijk, M.; Aten, J.; Overkleeft, H.S.; Balreira, A.; Zunke, F.; Schwake, M.; et al. Action myoclonus-renal failure syndrome: Diagnostic applications of activity-based probes and lipid analysis. J. Lipid Res. 2014, 55, 138–145. [Google Scholar] [CrossRef]
- De Boer, J.F.; Kuipers, F.; Groen, A.K. Cholesterol Transport Revisited: A New Turbo Mechanism to Drive Cholesterol Excretion. Trends Endocrinol. Metab. 2018, 29, 123–133. [Google Scholar] [CrossRef]
- Bietrix, F.; Lombardo, E.; Van Roomen, C.P.; Ottenhoff, R.; Vos, M.; Rensen, P.C.; Verhoeven, A.J.; Aerts, J.M.; Groen, A.K. Inhibition of Glycosphingolipid Synthesis Induces a Profound Reduction of Plasma Cholesterol and Inhibits Atherosclerosis Development in APOE*3 Leiden and Low-Density Lipoprotein Receptor−/− Mice. Arter. Thromb. Vasc. Biol. 2010, 30, 931–937. [Google Scholar] [CrossRef]
- Gamp, A.; Tanaka, Y.; Lullmann-Rauch, R.; Wittke, D.; D’Hooge, R.; De Deyn, P.; Moser, T.; Maier, H.; Hartmann, D.; Reiss, K.; et al. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum. Mol. Genet. 2003, 12, 631–646. [Google Scholar] [CrossRef]
- De Boer, O.J.; Van Der Meer, J.J.; Teeling, P.; Van Der Loos, C.M.; Idu, M.M.; Van Maldegem, F.; Aten, J.; Van Der Wal, A.C. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J. Pathol. 2009, 220, 499–508. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Donker-Koopman, W.E.; Van Der Vliet, M.K.; Jonsson, L.M.V.; Ginns, E.I.; Murray, G.J.; Barranger, J.A.; Tager, J.M.; Schram, A.W. The occurrence of two immunologically distinguishable beta-glucocerebrosidases in human spleen. Eur. J. Biol. Chem. 1985, 150, 565–574. [Google Scholar] [CrossRef]
- Witte, M.D.; Kallemeijn, W.W.; Aten, J.; Li, K.-Y.; Strijland, A.; E Donker-Koopman, W.; van den Nieuwendijk, A.M.C.H.; Bleijlevens, B.; Kramer, G.; I Florea, B.; et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Li, K.-Y.; Witte, M.D.; Marques, A.R.A.; Aten, J.; Scheij, S.; Jiang, J.; Willems, L.I.; Voorn-Brouwer, T.M.; Van Roomen, C.P.A.A.; et al. Novel Activity-Based Probes for Broad-Spectrum Profiling of Retaining β-Exoglucosidases In Situ and In Vivo. Angew. Chem. Int. Ed. 2012, 51, 12529–12533. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lienden, M.J.C.; Aten, J.; Marques, A.R.A.; Waas, I.S.E.; Larsen, P.W.B.; Claessen, N.; van der Wel, N.N.; Ottenhoff, R.; van Eijk, M.; Aerts, J.M.F.G. GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice. Int. J. Mol. Sci. 2021, 22, 2532. https://doi.org/10.3390/ijms22052532
van der Lienden MJC, Aten J, Marques ARA, Waas ISE, Larsen PWB, Claessen N, van der Wel NN, Ottenhoff R, van Eijk M, Aerts JMFG. GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice. International Journal of Molecular Sciences. 2021; 22(5):2532. https://doi.org/10.3390/ijms22052532
Chicago/Turabian Stylevan der Lienden, Martijn J. C., Jan Aten, André R. A. Marques, Ingeborg S. E. Waas, Per W. B. Larsen, Nike Claessen, Nicole N. van der Wel, Roelof Ottenhoff, Marco van Eijk, and Johannes M. F. G. Aerts. 2021. "GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice" International Journal of Molecular Sciences 22, no. 5: 2532. https://doi.org/10.3390/ijms22052532
APA Stylevan der Lienden, M. J. C., Aten, J., Marques, A. R. A., Waas, I. S. E., Larsen, P. W. B., Claessen, N., van der Wel, N. N., Ottenhoff, R., van Eijk, M., & Aerts, J. M. F. G. (2021). GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice. International Journal of Molecular Sciences, 22(5), 2532. https://doi.org/10.3390/ijms22052532