Abstract
Obesity is rapidly dispersing all around the world and is closely associated with a high risk of metabolic diseases such as insulin resistance, dyslipidemia, and nonalcoholic fatty liver disease (NAFLD), leading to carcinogenesis, especially hepatocellular carcinoma (HCC). It results from an imbalance between food intake and energy expenditure, leading to an excessive accumulation of adipose tissue (AT). Adipocytes play a substantial role in the tumor microenvironment through the secretion of several adipokines, affecting cancer progression, metastasis, and chemoresistance via diverse signaling pathways. AT is considered an endocrine organ owing to its ability to secrete adipokines, such as leptin, adiponectin, resistin, and a plethora of inflammatory cytokines, which modulate insulin sensitivity and trigger chronic low-grade inflammation in different organs. Even though the precise mechanisms are still unfolding, it is now established that the dysregulated secretion of adipokines by AT contributes to the development of obesity-related metabolic disorders. This review focuses on several obesity-associated adipokines and their impact on obesity-related metabolic diseases, subsequent metabolic complications, and progression to HCC, as well as their role as potential therapeutic targets. The field is rapidly developing, and further research is still required to fully understand the underlying mechanisms for the metabolic actions of adipokines and their role in obesity-associated HCC.
1. Introduction
Obesity is one of the most severe health disorders prevailing worldwide. Its prevalence has been increasing at an alarming rate for the last few decades. Obesity is defined as a body mass index (BMI) equal to or higher than 30 kg/m2. Obesity often leads to a series of medical disorders, including metabolic syndrome and nonalcoholic fatty liver disease (NAFLD), a spectrum including nonalcoholic steatohepatitis (NASH) [1]. It has also been recognized epidemiologically and clinically as a major risk factor for liver cancer. Because of its much wider spread and higher prevalence, obesity serves as a large contributor to overall hepatocellular carcinoma (HCC) [2]. The precise mechanisms underlying the obesity–liver cancer link are not yet completely unraveled. In general, obesity results from an impaired balance between calorie intake and energy expenditure, ultimately leading to the excessive accumulation of adipose tissue (AT). AT is not only recognized as a reservoir for excess energy derived from food but also as an endocrine organ. It produces adipocytokines or adipokines that trigger chronic low-grade inflammation in several organs of the body. It has been suggested that excessive AT and AT dysfunction dysregulate adipokine secretion, contributing to a variety of pathological processes, resulting in obesity-related liver cancer [3,4,5,6].
Broadly, AT is categorized into brown adipose tissue (BAT) and white adipose tissue (WAT). In the adult human body, the prevalence of BAT is minimal, only ~50 g in comparison to kilograms of WAT. Much evidence emphasizes that WAT is a metabolically active secretory endocrine organ [7,8]. It comprises different cells producing adipokines and cytokines [9]. Adipocytes tend to divide and give rise to new adipocytes upon stimulation and activation. The new white adipocytes increase or decrease in volume throughout their lifetime until the death of an individual [10].
WAT provides most of the total body fat, which is a source of free fatty acids (FFAs). FFAs are employed as substrates to generate energy via the oxidative phosphorylation of high-energy ATP bonds [11]. WAT is dispersed in the intra-abdominal region (omentum, intestines, and perirenal) and the subcutaneous region (buttocks, thighs, and abdomen) [11]. Two key functions of WAT are the regulation of metabolism and inflammation. It controls metabolism through energy homeostasis, adipocyte differentiation, and insulin sensitivity [12,13]. It regulates inflammation by the generation of pro- and anti-inflammatory molecules and the activation of metabolic and immune signaling [12,13]. Unfortunately, the excessive accumulation of WAT in body sites gives rise to obesity and obesity-associated diseases. In particular, WAT, deposited in the upper parts of the body, known as android obesity/central obesity, represents a risk factor for inflammatory pathologies [14]. Excess WAT deposited in the lower parts of the body give rise to gynoid obesity, with no metabolic issues [11,14]. Several theories have considered the underlying different distributions of WAT and its association with metabolic and inflammatory complications, including two theories that are not mutually exclusive. The first theory is based on the anatomical aspects of central obesity and its ability to drain FFAs and inflammatory mediators into the portal circulation, where they specifically act on hepatic cells, affecting the metabolism [11]. The second theory deals with the biological properties of WAT cells causing major/minor risks for developing metabolic and inflammatory complications [15]. Clinically, differential expression in several genes in different deposits of the body’s WAT has been reported [16,17]. The different types of cells that constitute WAT are mature adipocytes, preadipocytes, fibroblasts, endothelial cells, and macrophages [11,14,18]. Adipocytes, preadipocytes, and macrophages possess metabolic and inflammatory functions that cause WAT to release signaling molecules in WAT itself or other tissues at paracrine or endocrine levels [12,13,18,19]. Low-grade chronic obesity-related inflammation is specifically determined by macrophages that mediate circulating levels of specific inflammatory molecules [12,14,18,20,21]. Unlike WAT, BAT consists of a small number of fat cells with a rich vascular supply, an excess of mitochondrial chromogens, and energy expenditure from nonoxidative phosphorylation [22]. BAT has been highlighted as a potential target for pharmacological and genetic manipulation to combat obesity [23].
It is necessary to understand the process of energy homeostasis to interrogate the potential mechanisms involved in energy-rich conditions during overnutrition. The dynamic process of fat remodeling ensures adequate body fat and energy homeostasis without excess weight gain or loss. A positive energy balance with weight gain is dependent on conditions of increased appetite and food intake. Contrastingly, satiety determines a negative energy balance and weight loss by limiting food consumption. This whole phenomenon is under hypothalamic neuropeptide regulation of appetite and satiety [22]. Depending on physiological needs, a balance in energy homeostasis is achieved by the central and autonomic nervous system regulating energy expenditure [24]. Henceforth, more energy expenditure leads to WAT lipolysis and the augmentation of FFAs, whereas less energy expenditure favors an increase in fat storage [22]. The parasympathetic nervous system (PNS) enables fat deposition and a reduction in peripheral energy use [25]. The sympathetic nervous system (SNS) stimulates lipolysis by facilitating the release of FFAs for higher energy expenditure [26]. SNS innervations and β3 adrenergic receptors are predominant in both BAT and WAT. In BAT, uncoupled oxidative phosphorylation elevates heat production during cold-induced SNS activity [26,27]. In response to cold, high SNS activity stimulates WAT, which subsequently elevates thermogenesis through the oxidative phosphorylation of FFA in liver, muscle, and fat cells. This process is increased during obesity [26]. Heat-intolerant obese individuals tend to dissipate less heat from WAT and BAT, and at the same time, BAT mass and function are significantly reduced [22]. Thus, in general, obesity is induced by high calorie intake and reduced energy expenditure.
2. Lipid Metabolism in the Liver Regulated by Adipokines
Hepatic cells are mostly affected by ectopic lipid accumulation since the liver is the master regulator of systemic lipid and glucose accumulation. Consequently, NAFLD and NASH are the most common liver disorders, with up to a 90% prevalence in the obese population [28]. NAFLD and NASH are strongly associated with insulin resistance, progressing to cirrhosis and eventually to HCC, highlighting that excess lipid deposition in the liver leads to severe pathological consequences [29,30].
Albumin- and lipoprotein (LP)-mediated canonical lipid transport mechanisms regulate the mobility of lipids between tissues, such as the flux of lipids from AT to the liver and skeletal muscle or from the liver to AT and skeletal muscle. The impact of AT lipolysis on hepatic lipid accumulation has been illustrated by several studies, where fasting mice for 16 h induced nearly a 10-fold increase in hepatic lipid accumulation [31]. Several studies have identified mechanistic links, providing the basis for the AT–liver communication axis regulating systemic metabolic homeostasis and facilitating the understanding of the role of lipid transport, synthesis, and utilization in hepatic lipid metabolism and deposition [32,33,34]. Thus, it is important to understand the crosstalk between adipose tissue and the liver from a lipid-centric perspective in the maintenance of systemic energy homeostasis. Two AT-secreted factors, adiponectin (ADP) and leptin, play a seminal role in regulating lipid metabolism in the liver.
2.1. Adiponectin (ADP)
Serum ADP is found to be reduced in obese, insulin-resistant individuals [35]. ADP overexpression in obese conditions has been reported to prevent high-fat diet (HFD)-induced lipid accumulation [36]. Moreover, ADP knockout in leptin-deficient (ob/ob) obese mice augments hepatosteatosis [32]. The subtypes of adiponectin (ADP) receptors that are expressed in the liver are adipoR1 and adipoR2 [37]. ADP reduces hepatic lipogenesis and elevates fatty acid (FA) β-oxidation via the adipoR1-mediated activation of 5′-Adenosine Monophosphate-Activated Protein Kinase (AMPK) and Peroxisome Proliferators-Activated Receptor α (PPARα) [34,38,39]. AMPK inhibits lipogenesis by phosphorylating Acetyl-CoA Carboxylase-1 (ACC-1), reducing ACC-1 activity and malonyl CoA production thereby releasing the inhibition of Carnitine Palmitoyl Transferase 1 (CPT1) activity and stimulating the transport of FA into mitochondria for undergoing β-oxidation. At the transcriptional level, ADP induces the LKB-AMPK pathway via adipoR1, reducing the expression of hepatic lipogenesis and cholesterol synthesis genes by suppressing Sterol Regulatory Element Binding Protein 1c (SREBP1c) expression [34]. SREBP1c stimulates the expression of ACC-1 along with nuclear receptor Liver X Receptor (LXR) in the liver [40]. Hence, SREBP1c knockout prevents lipid accumulation in HFD-fed and leptin-deficient mice [41].
Independent of AMPK activation, the modulation of ceramide metabolism has been attributed to confer the pleiotropic functions of ADP, promoting insulin sensitivity, decreasing inflammation, and supporting cell survival [42]. ADP enhances ceramide catabolism by stimulating ceramidase activity through AdipoR1 and AdipoR2, resulting in the formation of the antiapoptotic molecule sphingosine-1-phosphate (S1P) [42]. The overexpression of acid ceramidase in HFD-fed mice prevents hepatic lipid accumulation and aids insulin sensitivity [33]. As such, ceramidase activity induced by ADP signaling might be key to its therapeutic efficacy. The metabolic effects of fibroblast growth factor 21 (FGF21), secreted by adipose tissue, the liver, and skeletal muscle, are facilitated by ADP. FGF21 has been reported to restore euglycemia, ameliorate hyperlipidemia, and reduce fat mass in obese models [43,44]. It was documented that FGF21 reduces ceramide accumulation in the liver and stimulates ADP secretion. Indeed, genetic ablation of ADP in leptin-deficient (ob/ob) and diet-induced obese mice prevented FGF21 from exerting its beneficial effects, indicating that FGF21 depends on ADP for lipid-lowering effects in obese mice [32].
2.2. Leptin
Leptin exhibits catabolic action in the adipocytes, preventing lipogenesis and activating β-oxidation of FA in the liver. Leptin receptors are abundantly expressed in the hepatic cells, and their expression is upregulated in response to leptin stimuli and short-term fasting [45]. Leptin-deficient ob/ob mice and leptin-receptor-deficient db/db mice have been reported to present with hypertriglyceridemia, hypercholesterolemia [46], and diminished lipid tolerance [47], ultimately leading to steatosis [48]. A study involving liver-specific leptin receptor knockout mice revealed reduced circulating levels of apolipoprotein B, high very-low-density lipoprotein triglyceride (VLDL TG), and hepatic lipoprotein lipase activity, suggesting changes in TG incorporation into VLDL or abnormal lipoprotein remodeling in the plasma [49]. Leptin also elevates hepatic FA oxidation and reduces de novo lipogenesis through ACC-1 phosphorylation [50].
Additionally, ADP and leptin exert anti-inflammatory actions on hepatic cells, aiding in the prevention of the transition from NAFLD to NASH. Antifibrotic and anti-inflammatory activity of ADP reduces hepatic fibrosis in steatohepatitis models [51,52]. Likewise, recombinant leptin and metreleptin (leptin analog), administered for lipodystrophy and congenital leptin deficiency, have been reported to reverse hepatic lipid accumulation and reduce the consequences of NASH [53]. Metreleptin is currently under clinical trial (NCT01679197), showing potential hope for adipose-derived secretory products to combat obesity-associated hepatic disorders.
3. Role of AT and Adipokines in Hepatic Functioning
AT comprises major cell types—adipocytes, preadipocytes, endothelial, and immune cells. It stores excess energy as TG in the lipid droplets of adipocytes through hyperplasia (increase in the number of adipocytes) or hypertrophy (size enlargement of adipocytes) [54]. The number of adipocytes is age-dependent, variable during childhood and adolescence, and constant during adulthood, irrespective of body weight (lean or obese) [55]. Elevated fat mass during adulthood primarily contributes to hypertrophy. A recent study has highlighted the hyperplasia of adipocytes in adulthood, where normal-weight adults expand only lower-body subcutaneous fat by increasing the number of adipocytes in response to overfeeding [56].
While obesity is allied with metabolic disorders, hypertrophy-mediated AT dysfunction plays a significant role in the development of insulin resistance (IR) [57]. During energy requirement, TGs stored in adipocytes are mobilized through lipolysis and release FFA, which subsequently move to other tissues to be employed as an energy source, as displayed in Figure 1.
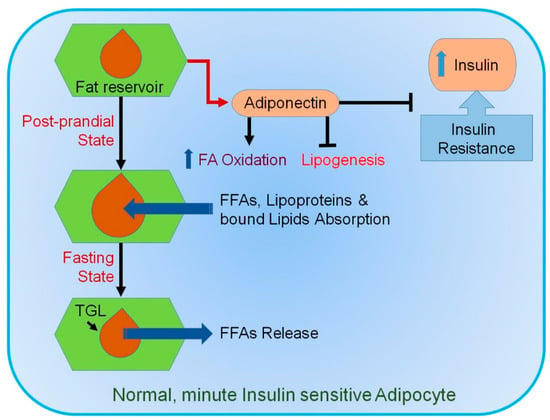
Figure 1.
Mechanism involved in normal healthy adipocytes. In response to a sedentary lifestyle, food intake, and an increase in circulating insulin, small healthy adipocytes absorb different types of lipids. They tend to suppress lipogenesis and secrete adiponectin (ADP). ADP promotes insulin sensitivity and fatty acid (FA) oxidation. During fasting, triglyceride lipases (TGLs) become activated, releasing free fatty acids (FFAs). Blue upward arrow indicates an increase.
FFAs play a vital role in the development of obesity-associated metabolic disorders. In obesity, FFAs enter the liver through the portal circulation, and high hepatic FFAs induce lipid synthesis, gluconeogenesis, and IR in the liver [58]. High levels of circulating FFAs leading to peripheral IR have been reported in both in vivo studies and in humans [58,59]. Additionally, FFAs modulate the inflammation of AT contributing to obesity-related metabolic complications by serving as ligands for the TLR4 complex [60] and stimulating cytokine production by macrophages [61]. Many studies also indicate a direct relationship between FFA release from AT and obesity-associated metabolic complications.
AT secretes multiple adipokines such as chemokines, cytokines, and hormones. Many of them are involved in energy homeostasis and inflammation. In obesity, adipocytes develop obesity-induced inflammation under the influence of a high secretion of chemokines and cytokines [62,63]. Many of them, such as MCP-1, TNF-α, IL-1, IL-6, and IL-8, promote IR [64,65,66,67]. Moreover, macrophages from AT are correlated with adipocyte size and body mass and proinflammatory cytokines [68]. Along with a high number of macrophages in AT, obesity stimulates a phenotypic switch from an anti-inflammatory M2 polarization state to a proinflammatory M1 polarization state [69]. The accumulated M1 macrophages in AT secrete a variety of proinflammatory cytokines and chemokines, contributing to obesity-linked IR [70]. On the other hand, M2-polarized macrophages facilitate AT remodeling, involving the clearance of dead adipocytes and the enrolment and differentiation of adipocyte progenitors [71]. The decreased expression of inflammatory cytokines in AT and increased insulin sensitivity in obese models have been observed by reduced adipose macrophage infiltration or macrophage removal [72,73]. Moreover, in obese conditions, weight loss reduces macrophage infiltration and proinflammatory gene expression in adipose tissue [74,75]. In obesity, proinflammatory immune cells, such as interferon (IFN)-γ+T helper type 1 cells and CD8+T cells, are elevated [76], with reduced secretion of insulin-sensitizing adiponectin in AT [77].
WAT produces and secretes adipocytokines or adipokines, but in hypertrophy, a change in protein synthesis leads to the production of inflammatory proteins, such as cytokines [78,79,80]. In general, adipokines, such as TNF-α, IL-6, and PAI-1, are proinflammatory, whereas adiponectin is anti-inflammatory, antidiabetic, cardioprotective, and antitumorigenic [81,82]. In obesity, dysfunctional AT releases higher levels of proinflammatory factors and reduced adiponectin [83]. This difference in the pathophysiology of adipocytes aids in understanding the association between obesity, IR, metabolic syndrome, atherosclerosis, and cancer [84,85]. A close association exists between the production of inflammatory proteins and the level of hypertrophy of adipocytes [61,86]. A summary of key proteins secreted by adipocytes normally and during hypertrophy is shown in Table 1.

Table 1.
List of proteins secreted by adipocytes during normal conditions and hypertrophy.
Adipokines comprise heterogeneous proteins, which, functionally, are polyvalent molecules involved in physiological and pathological processes modulating the sensitivity of peripheral tissues to insulin, regulating appetite, homeostasis, energy expenditure, and glucose and lipid metabolism [87,88]. Additionally, they are strongly related to immunity and inflammation [89,90]. WAT plays a crucial role in controlling physiological and pathological processes (metabolism and energy homeostasis) [91,92]. It communicates directly with peripheral tissues, particularly skeletal muscle, through various adipokines. Intense crosstalk exists between WAT and the brain through leptin and SNS [93,94].
Plasma TNF-α levels are higher in hyperlipidemic patients and are positively correlated with VLDL TG concentrations [95], which are associated with hepatic TG synthesis and the secretion and inhibition of LPL [96,97]. Additionally, TNF-α promotes the synthesis of hepatic apolipoprotein (apo) B100-VLDL by impairing hepatic insulin signaling [98]. Similarly, IL-6 is also related to hypertriglyceridemia. Hypertriglyceridemia and upregulated serum TG subjects produce high levels of IL-6 and TNF-α [99,100]. Interestingly, high anti-inflammatory cytokine levels, such as IL-10, are also related to high-plasma TG levels [101]. Other proinflammatory cytokines, IL-6, IL-1, IFN-α, and IFN-γ, promote TG synthesis in HepG2 cells [102], stimulate lipolysis in adipocytes [103,104], and reduce LPL activity in vivo and in vitro [105,106,107]. Many studies also report that TNF-α and IL-6 are negatively related to serum HDL-cholesterol levels in healthy individuals and cardiovascular patients [108,109], and IL-10 is positively related to plasma HDL-cholesterol levels [101]. There are also reports of the TNF-α-, IL-6-, and IL-1-mediated reduced expression of apo A1 in hepatic cells and plasma [105]. Proinflammatory cytokines upregulate circulating total cholesterol and LDL-cholesterol levels by activating cholesterol synthesis [105,110], and higher IL-10 levels are negatively correlated with increased levels of total cholesterol and LDL [101]. TNF-α, TGF-β, or IL-1 promote lipoprotein uptake by LDL and scavenger receptors and inhibit ABCA1-mediated cholesterol efflux to HDL, which contributes to lipid deposition and the formation of foam cells [111,112,113]. Additionally, in the absence of extracellular FAs, TNF-α increases the secretion of apo B in hepatocytes [114]. Cytokine treatment also stimulates the hepatic production and secretion of phospholipase A2 [115].
4. Adipokine-Mediated Modulation of Cell Death and Survival in Hepatocytes
4.1. Prosurvival Effects of ADP
ADP contributes toward effects on metabolic disorders and the development and progression of cancer. It has been reported to suppress tumor growth and inversely associate with the incidence of cancer [116]. ADP modulates hepatic physiology by controlling cell death and the survival of primary hepatic cells [117]. ADP protects hepatic cells from iron-overload-induced hepatic injury by Heme Oxygenase 1 (HO1) induction and PPAR-α activation [118] and suppresses ethanol-induced apoptotic cell death through Nrf-2 signaling and HO1 induction [119]. Hepatic apoptosis is considered to be an early feature of alcoholic liver disorder. ADP protects hepatic cells by inhibiting CD95 upregulation [120] and ameliorates AMPK/eNOS signaling-mediated hepatic apoptosis and inflammation [121], thereby providing a prominent therapeutic option for alcohol-induced liver injury. It has been shown to induce autophagy and promote cell survival by enhancing autophagy-related gene expression through the AMPK-dependent nuclear translocation of FoxO3A [119]. ADP-mediated autophagy digests Bax and prevents caspase activation [119], and adipoR1 signaling enhances the expression of chemokine-like receptor 1 and CXCL8 [122,123] and attenuates ceramide accumulation. AdipoR1 and R2 signaling increases ceramidase activity and the formation of S1P, which contributes to the prosurvival effect of ADP in the liver [42].
Hepatocyte Growth Factor (HGF) and FGF-2 produced by hepatic stellate cells (HSCs) promote hepatic fibrosis [124,125]. ADP induces apoptosis and inhibits HSCs, affects the survival of Kupffer cells, and promotes conversion to the M2 phenotype for releasing mediators that stimulate M1 macrophage apoptosis [126]. Additionally, it induces apoptosis of murine macrophage cells through reactive oxygen species (ROS) production from reduced NADPH oxidase and iNOS [127,128], affects activated and/or quiescent status of HSCs, prevents the proliferation of HSCs, and induces apoptosis of activated HSCs [129]. ADP induces the expression of p53 and Bax and inhibits the GSK-3β mediated prosurvival pathway, thereby inhibiting the activity of β-catenin or cyclin D1.
4.2. Effect of Leptin on Proliferative Potential of Hepatic Cells
Leptin mainly induces the proliferation and activation of HSCs without exerting significant proliferation/survival effects on hepatocytes and Kupffer cells. It is predominantly produced from AT and activated HSCs, impairing hepatic fibrosis resolution [129], inhibiting TRAIL-induced apoptosis, and suppressing FasL-induced apoptosis in HSCs [130]. Leptin also promotes liver fibrosis by inducing Transforming Growth Factor β (TGF-β) and connective tissue growth factor in HSCs [131]. The leptin receptors expressed in hepatocytes are involved in ROS production through NADPH oxidase-dependent signaling, although it does not significantly affect the function of primary mouse hepatocytes [132]. However, leptin has been reported to stimulate proliferation and suppress apoptosis of hepatic cancer cells. Its plasma level is higher in obesity, and its role in cancer progression suggests an underlying mechanism for cancer development in obese individuals [133]. It induces cell cycle progression and elevates the number of cells in the S and G2-M phase, enhancing DNA synthesis and mitotic activities [134]. The inhibition of the ER stress-associated apoptotic pathway contributes to a leptin-induced increase in liver cancer cells [135]. The induction of methionine adenosyltransferases confers the mitogenic property of leptin in liver cancer cells [136]. Leptin induces autophagy via a p53/Foxo3A axis, which, in turn, abrogates apoptosis of cancer cells [137].
Apoptosis in hepatic cells has been reported to mediate NAFLD/NASH and stimulate immune cells and hepatic stellate cells to produce inflammasomes and cytokines, leading to liver fibrosis [138]. These processes are usually accelerated by microbiota and abnormal glucose and lipid metabolism. In NASH, saturated FAs and mitochondrial cholesterol accumulation induce hepatic apoptosis, which involves the production of reactive oxygen species, oxidative stress, and endoplasmic reticulum stress [138]. Apoptotic events in NASH are caspase-dependent and are associated with mitochondrial membrane depolarization and cytochrome c release. Subsequently, the mitochondrial apoptotic pathways are activated through the activation of Bax and Bim, and c-Jun N-terminal kinase (JNK) plays an important role in mediating these events [139,140]. In the liver, MLK3 is one of the MAP3K proteins that mediate JNK activation during NASH [141]. Apoptosis inhibitors have been employed for NASH treatment, cirrhosis prevention, and HCC. A list of important apoptotic markers mediating NAFLD/NASH is shown in Table 2.
Table 2.
Different apoptosis markers involved in nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH).
Table 2.
Different apoptosis markers involved in nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH).
| Type | Subtype | Effect | Case | References |
|---|---|---|---|---|
| Caspases | Caspase 3 and 7 | Strongly correlated with hepatocyte apoptosis | NASH | [142] |
| Correlated with disease severity | NAFLD | |||
| Caspase 3 generated CK-18 fragments | NASH Predictor | [142,143] | ||
| Caspase 9 | Executes mitochondrial apoptosis pathway | NASH | [144] | |
| Caspase 2 | Initiator caspase in lipid-induced cytotoxicity | NASH | [145] | |
| Bcl-2 | Bax | NAFLD, NASH | [146] | |
| Bcl-2 | Promotes apoptosis by modifying the expression and function of Bcl-2 homology 3 (BH3) only protein Bim and PUMA | Hepatic lipoapoptosis | [147] | |
| Transglutaminase 2 | Crosslinks and inactivates transcription factor Sp1, resulting in hepatic apoptosis | NASH | [148] | |
| Bid | Higher expression of HDMCP induces hepatocyte apoptosis triggered hepatic stellate cell activation. | NASH | [149] | |
| JNK | Sh3bp5 | Impaired respiration, ROS production, JNK activation, and apoptosis | NASH | [150] |
| CREG | Interact with ASK1 and inactivate ASK1-JNK1 signaling | NAFLD | [151] |
5. Putative Mechanisms for Obesity-Induced Liver Cancer
5.1. Function of Adipose-Tissue-Produced Adipokines in Insulin Resistance
Insulin resistance (IR), defined by decreased responsiveness to insulin and characterized by reduced glucose disposal in nonhepatic tissues, is one of the obesity-related disorders posing a major risk toward cancer development and is associated with poor prognosis [152]. IR patients exhibit high glucose and insulin levels due to reduced insulin sensitivity [153]. The dysfunction of adipocytes, either because of genetic factors or induced by HFD, plays an integral role in IR [154]. WAT synthesizes, esterifies, and stores TG in lipid droplets, and it is highly insulin-sensitive [155]. Insulin acts by binding to the insulin receptor (IR) with the activation of the PI3K/Akt pathway and increases glucose uptake, augments the utilization of glucose for the production of glycerol, and inhibits lipolysis by lipases [155]. In obesity, IR promoted by proinflammatory cytokines leads to the chronic low-grade inflammation of AT, thereby providing a microenvironment for tumorigenesis [156]. Hypoxic hypertrophic adipocytes have elevated levels of inflammatory cytokines (TNF-α, IL-1, and IL-6) and reduced ADP [157]. These cytokines can function either directly by deactivating insulin receptor substrate-1 (IRS1) via the phosphorylation of serine and threonine residues or decreased tyrosine phosphorylation [158,159,160,161] or indirectly, wherein upregulated FFA increases NF-kB, which is implicated in IR [162].
5.1.1. Chemokines
In obesity, the expression of chemokines and their receptors is augmented in visceral and subcutaneous adipose tissue. Chemokines selectively recruit monocytes, neutrophils, and lymphocytes and induce chemotaxis [163]. C-C motif chemokine ligand 2/macrophage chemoattractant protein-1 (CCL2/MCP-1) regulates the migratory and infiltrative potential of monocytes/macrophages and initiates the inflammation of AT [164]. It is expressed by adipocytes and is correlated with adiposity. It has been reported that in AT, the overexpression of CCL2/MCP-1 elevates macrophage recruitment [165], whereas knockdown reduces the accumulation of proinflammatory macrophages, providing protection from IR and hepatic steatosis [68,165]. Interestingly, primary adipocytes show inherent immune function in which adipocyte-derived CCL2/MCP-1 stimulates inflammation and activates CD4+ T cells independently of macrophages/leukocytes in AT [166].
Other chemokines involved in AT macrophage infiltration and obesity-induced IR are CCL5, C-X-C motif chemokine ligand 5 (CXCL5), and CXCL14 [73,167,168]. In AT of obese individuals, multiple chemokines, CCL2, CCL3, CCL5, CCL7, CCL8, CCL11, CCL19, CXCL1, CXCL5, CXCL8, and CXCL10, and their receptors, CCR1, CCR2, CCR3, and CCR5, are highly expressed [163,168]. It has been shown that CXCL5, secreted from WAT-resident macrophages, interferes with insulin function by activating Jak2/STAT5/SOCS2 signaling, thus inducing IR, and treatment with anti-CXCL5 neutralizing antibody or antagonists of CXCL5 receptor CXCR2 provides protection in obese, IR mice [168].
5.1.2. TNF-α
TNF-α is highly expressed in obese and IR individuals and is positively correlated with IR [169]. Indeed, TNF-α deletion improves insulin sensitivity in obese mice [170]. In healthy overweight individuals with metabolic syndrome and IR, the correlation between plasma TNF-α levels and IR is rather weak [64,171], and the chronic neutralization of TNF-α fails to improve the status of IR [172], suggesting an obesity-specific function of TNF-α. It has been also reported that TNF-α antagonist etanercept administration fails to improve insulin sensitivity in patients with metabolic syndromes [173], which might be explained by the compensatory role of other cytokines in the absence of TNF-α since various other proinflammatory cytokines, such as IL-1, and IL-6 secreted by AT, are involved in the disruption of insulin signaling [174].
5.1.3. IL-6 and IL-18
IL-6 plays a crucial role in developing IR in obesity [175]. AT contributes to 10–35% of circulating IL-6 levels, and the hypertrophic enlargement of adipocytes results in high levels of IL-6 [176,177], which are positively correlated with IR [178]. IL-18 is a TNF-α-induced proinflammatory cytokine produced by AT in high levels in obese individuals, and its levels are reduced with weight loss [179]. The overexpression of IL-18 aggravates IR in rats [180]. Interestingly, the deletion of IL-18 or its receptor in mice induced hyperphagia, obesity, and IR because of increased food intake [181]. The lack of IL-18 in the liver caused the defective phosphorylation of STAT3, resulting in the enhanced expression of genes regulating gluconeogenesis contributing to hepatic IR. The intracerebral administration of recombinant IL-18 inhibited food intake and reversed hyperglycemia, indicating the tissue-specific function of IL-18 in AT and in the brain [181].
5.1.4. Leptin
Leptin is profusely expressed in adipocytes regulating energy and glucose homeostasis, inhibiting appetite and food intake and stimulating energy expenditure [182]. A decrease in fat mass results in the lowering of plasma leptin levels, stimulating appetite and suppressing energy expenditure, while in obesity, increased fat mass results in increased leptin levels, suppressing appetite until body weight is decreased. Circulating leptin levels and leptin levels in AT are proportional to fat mass, but the level is rapidly decreased by fasting and increased by proinflammatory cytokines [183,184,185,186]. A bidirectional interaction exists between leptin and inflammation, in which proinflammatory cytokines elevate the synthesis and release of leptin, contributing to a chronic inflammatory state in obesity [187]. In the liver and skeletal muscle, leptin improves insulin sensitivity, but it exerts tissue-specific effects so that it inhibits adipocyte insulin signaling as an autocrine signal by abrogating insulin-induced MAPK activation, GSK3β phosphorylation, and insulin receptor tyrosine phosphorylation. Furthermore, indirectly through neuroendocrine pathways and in pancreatic β-cell, it impairs insulin secretion [188,189]. Leptin’s cytokine-like structure and its receptor, a member of the class I cytokine receptor (gp130) superfamily, promotes the production of proinflammatory (Th1) cytokines (IL-2 and IFN-γ) and inhibits the production of anti-inflammatory (Th2) cytokine (IL-4) by T cells/mononuclear cells, thus exerting an immunomodulatory effect [187,190].
5.1.5. Resistin
Resistin is an adipokine promoting inflammation and IR [191]. Circulating levels of resistin are higher in obesity and are correlated with IR. Hyperresistinemia created by acute resistin infusion or stable resistin gene transfer causes IR in rodents, whereas its absence protects from diet-induced hyperglycemia as well as IR in ob/ob mice by increasing AMPK activity and decreasing the levels of gluconeogenic enzymes in the liver [192,193,194]. It inhibits multiple steps in insulin signaling, including IR and IRS-1 phosphorylation, PI3K activation, phosphatidylinositol triphosphate production, and the activation of Akt, by inducing the expression of an inhibitor of insulin signaling suppressor of cytokine signaling-3 (SOCS-3) in adipocytes and AT [195]. In rodents, resistin is produced exclusively from AT, whereas in humans, resistin is produced mainly by mononuclear cells, especially macrophages, and macrophage-derived human resistin exacerbates AT inflammation and IR [196,197].
5.1.6. PAI-1
A primary inhibitor of fibrinolysis PAI-1 is synthesized in AT by adipocytes and stromal vascular cells, such as preadipocytes, fibroblasts, vascular endothelial cells, and immune cells. Its levels are high during obesity and IR [198,199]. The lack of PAI-1 reduces body weight, increases total energy expenditure, improves IR in high-fat diet-fed mice by increasing uncoupling protein 3 mRNA expression in skeletal muscle and maintaining PPARγ and adiponectin levels in WAT [200], and promotes adipocyte differentiation, improving basal glucose and insulin-stimulated glucose uptake [201]. It regulates the expression of IL-8 and leukotriene B4 and monocyte migration under the influence of cytokine inducers such as cigarette smoke extraction and LPS [202].
5.1.7. Visfatin
Visfatin is a β-cell differentiation modulator expressed in lymphocytes, bone marrow, muscle, liver [203], and AT (especially visceral AT) [204]. It is a NAD biosynthetic enzyme, the haplodeficiency or chemical inhibition of which cause defects in NAD biosynthesis and glucose-stimulated insulin secretion [205]. Visfatin activates the insulin receptor and ERK1/2 signaling, thus improving glucose uptake in adipocytes and myocytes [206]. However, several studies report that circulating visfatin levels are high in obese individuals and that visfatin is an AT proinflammatory molecule associated with systemic IR and hyperlipidemia [207,208]. Serum visfatin levels have been shown to positively associate with serum IL-6 and C-reactive protein but not with IR [209]. The role of visfatin, therefore, is still a debatable issue and requires in-depth experimentation for clarification.
5.1.8. Retinol Binding Protein 4 (RBP4)
RBP4 is a hepatocyte-synthesized protein that aids in vitamin A transport in the body [210]. Additionally, it is secreted by adipocytes that affect insulin sensitivity [211]. RBP4 levels are elevated in insulin-resistant mice and humans with obesity and type 2 DM. The injection of recombinant RBP4 or the transgenic overexpression of human RBP4 in normal mice causes IR [211]. RBP4 is produced by visceral adipose tissue in states of obesity and IR [212]. In insulin-resistant mice, RBP4 expression is high in AT, and AT RBP4 mRNA expression is correlated with serum RBP4 levels [212]. RBP4 inhibits the insulin-induced phosphorylation of IRS-1 and ERK1/2 in primary adipocytes [213]. Apart from markers of obesity and IR, RBP4 is also correlated with inflammatory factors [214].
5.1.9. Angiopoietin-Like 2 (ANGPTL2)
ANGPTL2 is an adipocyte-derived inflammatory mediator promoting adiposity, inflammation and IR, and it is highly expressed in obese states in both mice and humans [215]. Its overexpression in AT promotes inflammation and IR in nonobese mice, whereas its deficiency ameliorates AT inflammation and IR in diet-induced obese mice. It activates integrin signaling to induce an inflammatory cascade and promotes chemotaxis of monocytes and macrophages [215].
5.1.10. Secreted Frizzled-Related Protein 5 (SFRP5)
SFRP5, a protein linked to the Wnt signaling pathway, is an insulin-sensitizing and anti-inflammatory adipokine expressed at higher levels in AT, exhibiting beneficial effects on metabolic dysfunction [216]. Its deficiency in high-calorie diet-fed mice exhibited impaired insulin sensitivity, increased risk for NAFLD and adipose inflammation, and the accumulation of macrophages and proinflammatory cytokines in AT by activating JNK1. Conversely, SFRP5 administration enhanced metabolic function and reduced adipose inflammation in obese states [216]. Circulating SFRP5 was decreased in children with obesity and metabolic syndrome and, upon weight loss, its levels showed an increase [217].
5.2. Function of Adipose Tissue-Produced Adipokines in Dyslipidemia
High levels of FFA released from AT through increased lipolysis because of IR are delivered to the liver. The enhanced FFA increases TG and VLDL production in the liver. The inhibition of lipases in AT and skeletal muscle and increased VLDL in the liver inhibiting the lipolysis of chylomicrons contribute to hypertriglyceridemia. The TG in VLDL produces TG-rich LDL and HDL through an exchange for cholesteryl esters from LDL and HDL via cholesteryl ester transport protein. The hepatic lipase hydrolyzes TG in LDL and HDL to both small, dense LDL and HDL. The reduced HDL concentration and formation of small, dense LDL are highly associated with metabolic diseases.
Cytokines and Serum Amyloid A (SAA)
TNF-α has been initially recognized to induce hypertriglyceridemia. Hyperlipidemic patients with higher levels of plasma TNF-α are positively correlated with VLDL TG concentration [95], promoting hepatic TG synthesis and secretion [96] and LPL inhibition [97]. Additionally, TNF-α promotes the synthesis of hepatic apolipoprotein (apo) B100-containing VLDL by impairing hepatic insulin signaling [98]. IL-6 is also associated with hypertriglyceridemia and the anti-inflammatory cytokine. IL-10 levels showed an inverse correlation with increased TG, total cholesterol and LDL-cholesterol levels, and metabolic syndrome [101,218]. Cytokines, such as TNF-α, IL-1, and IFN-α, have been reported to stimulate TG synthesis [102] and promote lipolysis in adipocytes [103,104]. The combination of TNF-α and IL-1 increased serum cholesterol, decreased HDL, increased hepatic HMG CoA reductase, and decreased hepatic apo E or apo A–I mRNAs [105]. TNF-α, TGF-β, or IL-1 contribute to lipid deposition and foam cell formation by promoting lipoprotein uptake by the scavenger receptor and the LDL receptor and inhibiting ABCA1-mediated cholesterol efflux to HDL [111,112,113].
SAA proteins, found in the liver and AT and markedly elevated in acute phase response, are expressed highly in obese subjects [219,220]. SAA also play a role in the inflammatory process. SAA treatment increases the expression of IL-6 and TNF-α in preadipocytes and adipocytes [221,222]. Additionally, SAA affect HDL-cholesterol metabolism by inhibiting scavenger receptor SR-BI-mediated HDL binding and selective lipid uptake [223]. SR-BI mediates the cellular uptake of cholesteryl esters from HDL, ultimately promoting the reversal of cholesterol transport from the periphery to the liver [224]. The increased expression of SAA promotes dyslipidemia by affecting HDL structure and function as well as inflammation [225].
5.3. Function of AT-Produced Adipokines in NAFLD
The most common form of chronic liver disease is NAFLD [226], and its incidence increases parallelly with a rise in the incidence of obesity [30]. More than two-thirds of NAFLD patients are obese [227]. The two steps of liver injury involved in NAFLD are, firstly, the accumulation of TG in the liver (hepatic steatosis) and, secondly, inflammation and subsequent fibrosis (nonalcoholic steatohepatitis, NASH) [228]. The “two-hit” hypothesis explaining the development of NAFLD and the progression from simple steatosis to NASH is the “first hit,” the accumulation of hepatic lipids, and the “second hit” promotes hepatocyte injury, inflammation, and fibrosis. The second hit is under the influence of proinflammatory cytokines, adipokines, mitochondrial dysfunction, oxidative stress, and subsequent lipid peroxidation [229].
Hyperinsulinemia results in steatosis through increased de novo hepatic lipogenesis and efflux of FFA due to high lipolysis from AT and decreased FFA oxidation and hepatic VLDL secretion. The influx of lipids induces excessive lipid accumulation through high FA import or de novo FA synthesis, overriding hepatic lipid clearance by FA oxidation or the export of TG [230,231]. Insulin failure in suppressing lipolysis results in the secretion of FFA from AT. Elevated lipolysis in AT increases FFA influx into the liver through the portal vein [232]. FFA from enlarged AT are then taken up by the hepatocytes, and a reduction in hepatic insulin clearance further increases circulating insulin levels [233]. The contribution of visceral AT lipolysis to the delivery of FFA is only 5–10% in normal-weight subjects and up to 25% in intra-abdominally obese subjects [234]. After developing steatosis, the liver becomes more prone to “multihit,” such as bacterial toxins, an imbalance in adipokine/cytokine, mitochondrial dysfunction, oxidative damage, impaired hepatocyte apoptosis, profibrogenic factors, and proinflammatory mediators, released from dysregulated organelles and the activated hepatic stellate cell and Kupffer cells. These multiple factors stimulate inflammation, apoptosis, and fibrosis, ultimately leading to progressive liver disease. Macrophage infiltration and the secretion of proinflammatory chemokines, cytokines, and adipokines are promoted by expanded AT, which is closely related to IR [68].
ADP protects the liver from steatosis and inflammation and increases the ability of insulin to suppress glucose production and glucose output [235]. It also inhibits hepatic lipogenesis by suppressing lipogenic transcription factor SREBP1-c [34] and promotes glucose utilization and FA oxidation in the liver by AMPK activation [38]. Circulating ADP levels are low in NAFLD individuals [235] and negatively correlated with liver function markers in healthy individuals [236]. Hepatic steatosis and increased liver injury enzyme levels are predicted by low ADP levels in obese individuals [237]. Additionally, ADP and AdipoR2 expression are reduced in NASH patients [238].
Leptin is another key regulator of NAFLD that stimulates AMPK involved in the activation of lipid oxidation and the inhibition of lipogenesis [239]. However, leptin has distinct roles in different hepatic cells. Leptin function in hepatocytes protects from hepatic steatosis and protects from hepatic injury, whereas it activates HSCs with the production of α-smooth muscle actin, collagen, and TIMP-1, leading to hepatic fibrosis, and upregulates the expression of TGF-β1 in Kupffer cells [240,241,242]. A clinical study demonstrated a positive correlation between circulating leptin and high-serum ALT or hepatic steatosis independent of BMI and body fat, although the significance of this finding is not clear [243].
Among the cytokines, NAFLD patients show increased circulating levels of resistin [244,245], and TNF-α not only mediates the early stages of NAFLD but also transitions to more advanced stages of liver damage [246,247,248]. Acylation-stimulating protein (ASP) and angiotensinogen levels expressed in AT are generally higher in obese subjects [249,250,251]. In NAFLD, the levels of ASP are reported to be associated with IR [249] and angiotensin II antagonists have been shown to improve liver function test and attenuate fibrosis [252].
5.4. Role of BCAA as a Mediator of Obesity and NAFLD
Branched-chain amino acids (BCAAs), a group of essential amino acids (leucine, isoleucine, and valine) with nonlinear aliphatic side-chains, are relatively abundant in food, accounting for 20% of total protein intake [253]. They are considered contributors to the development of obesity-associated IR because of the chronic phosphorylation of mTOR, JNK, and IRS1Ser307 and the accumulation of multiple acylcarnitines in muscle [254,255,256]. Levels of fasting plasma BCAAs are found to be elevated in obesity-associated NAFLD [256,257], coinciding with abnormalities in hepatic and AT BCAA catabolic enzymes [258], thus reducing the adipose browning mechanisms. Such malfunctioning in the hepatic BCAA metabolism affects carbon substrate oxidation, impaired antioxidant defense, and ROS generation [254,259]. NAFLD-associated mitochondrial dysfunction (hepatic inflammation) conveys impaired BCAA metabolism, leading to elevated plasma levels of BCAA [260].
A dual effect of BCAA on adipocyte lipolysis and hepatic lipogenesis has been described using HFD and HFD+BCAA mouse models [261]. Although BCAA decreased HFD-induced weight gain and inhibited hepatic lipogenic enzymes, leading to decreased TG content, it induced hepatic damage by activating mTOR, inhibiting hepatic autophagy and increasing hepatic oxidative stress and apoptosis. BCAA increased plasma FFA levels by increasing AMPK-mediated adipocyte lipolysis, and inhibiting lipolysis normalized plasma FFA levels and improved insulin sensitivity but did not protect from BCAA-induced hepatic damage. Thus, BCAA may exacerbate hepatic lipotoxicity [261].
6. Therapeutic Implications to Target Adipokines in Obesity-Induced Liver Cancer Progression
The correlation between obesity and cancer is majorly significant because of the convergent pathways involving adipokines, inflammation, and IR. Therapeutic intervention to prevent the effect of obesity on cancer is still under intense scrutiny. The study of bariatric surgery data shows an association between weight loss postsurgery and reduced cancer incidence and metastasis [262]. Patients on insulin or insulin-secreting agents (sulphonylureas) are at higher risks of cancer than patients on oral insulin-sensitizing agents (metformin or thiazolidinediones) [263,264]. Metformin and TZDs have been reported for antitumorigenic effects either as a single agent or in combination with conventional chemotherapeutic agents because of the reduction of insulin and the modulation of intracellular drug targets and epidemiologically both are reported to be associated with low cancer incidence rates [265,266,267]. However, their effect on obese patients with cancer is still not clear. Another potential therapeutic strategy of obesity-induced cancer progression is the chemopreventive modulation of inflammatory pathways.
6.1. ADP Agonists
ADP has been identified as a potential therapeutic and preventive agent in altering cancer outcomes, biology, and anticancer immunity. However, the pleiotropic nature of various ADP isoforms poses hurdles in engineering an effective ADP analog [268]. The first ADP receptor agonist with an acceptable safety profile reported was ADP355, which binds both ADP receptors, regulating AMPK, STAT3, PIK3/Akt, and ERK1/2 signaling pathways [268]. ADP355 inhibited the growth of tumor xenografts and protected from CCl4-induced liver fibrosis and thioacetamide-induced liver injury [268,269,270,271]. A fluorescence polarization-based screening of 10,000 natural compounds identified AdipoR1 agonists, such as matairesinol, arctiin, (–)-arctigenin, and gramine, as well as AdipoR2 agonists, such as parthenolide, taxifoliol, deoxyschizandrin, and syringin [272]. AdipoRon, an orally active synthetic small molecule AdipoR agonist, which activates both AdipoR1 and AdipoR2, activates AMPK and PPAR-α signaling and ameliorated IR and glucose intolerance in mice fed a HFD [273]. However, these agonists have not been tested for their efficacy in inhibiting obesity-associated liver cancer. In a phase I clinical trial, Efatutazone, a highly selective agonist of PPARγ, in patients with solid cancers showed a dose-dependent increase in plasma ADP levels and delayed invasive progression of mammary carcinogenesis [274,275]. PPARγ agonists, TZDs, induced ADP expression and secretion in a dose- and time-dependent manner in humans and rodents in vitro and in vivo and suppressed the growth, migration, and invasion of cancer cells by augmenting ADP expression and inhibiting leptin signaling [276,277]. As insulin sensitizers, both TZDs and metformin have shown efficacy in rodent models of NASH-HCC and metformin treatment showed a small but significant improvement in survival in patients with type 2 DM and HCC in a clinical study [278,279,280].
6.2. Leptin Inhibitors
Leptin exerts pleiotropic functions so that, on the one hand, it increases insulin sensitivity in hepatocytes and, on the other, it promotes hepatic fibrosis via HSCs and can directly stimulate proliferation and antiapoptosis in cancer cells to augment cancer progression. Both leptin and its receptor are overexpressed in HCC. Leptin activates oncogenic signaling pathways, such as PI3K/Akt, MAPK, and STAT3 and upregulates human telomerase reverse transcriptase (hTERT), a mediator of cellular immortalization, in HCC cells [281,282]. Leptin inhibition has been tested as a therapeutic strategy mostly in breast cancer. According to the leptin binding site, leptin peptide receptor antagonist (LPrA) comprises LprA1 and LprA2. LprA2 is conjugated to polyethylene glycol 20kDa (PEG-LprA2) or iron-oxide nanoparticles. PEG-LprA2 inhibits breast cancer by interfering with the expression of proangiogenic factors (VEGF/VEGFR-2) and proproliferative factors (PCNA, cyclin D1) [283]. The in vivo efficacy of PEG-LPrA2 was more significant because of paracrine signaling between stromal, endothelial, and/or inflammatory cells, which play an important role in cancer progression. LPrA2, conjugated with iron-oxide nanoparticles, reduced leptin-induced chemoresistance for cisplatin, cyclophosphamide, paclitaxel, and doxorubicin in breast cancer [284]. LDFI, a leptin receptor antagonist peptide, suppressed the growth of breast cancer cells by inhibiting leptin downstream molecules, such as JAK2, STAT3, AKT, and MAPK [285]. A JAK2 inhibitor AG490 inhibits STAT3 phosphorylation, cell proliferation, HER2 transphosphorylation, and HER2 stabilization by suppressing leptin/Jak2/STAT3 signaling [286,287,288]. Recently, in vitro studies with synthetic leptin derivatives demonstrated inhibition of the proliferation of HCC cells [289]. However, considering the role leptin plays in IR, fibrosis, and HCC, carefully monitored controlled studies with appropriate clinically relevant models need to be performed to use leptin inhibitors as a potential therapy for obesity-induced HCC.
6.3. Autotaxin-Lysophosphatidic Acid (ATX-LPA) Signaling Inhibitors
ATX is a secreted enzyme that converts lysophosphatidyl choline into LPA, which signals through LPA receptors to promote hepatic fibrosis and cancer [290]. AT produces ~35% of the body’s ATX, and ATX production is increased during obesity [291]. An autotaxin inhibitor PAT-505 reduced fibrosis in mouse models of NASH and an ATX inhibitor AM063 and an LPAR1 antagonist AM095 decreased fibrosis and reduced HCC development in a diethylnitrosamine (DEN)-induced HCC model [292,293]. Hepatocyte-specific ATX-deficient mice are protected from fibrosis and HCC, thus further establishing the importance of this pathway [294].
6.4. IL-6 Inhibitors
Blocking IL-6 signaling might serve as an effective therapeutic strategy for cancer [295]. IL-6 signaling might be inhibited in several ways, such as IL-6-conjugated toxins or IL-6 monoclonal antibodies. A monoclonal antibody, CNTO 328 (siltuximab) inhibits JAK/STAT3 signaling and IL-6/IL-6R/gp130 trans-signaling by interfering with the binding of IL-6 and IL-6R and has shown encouraging results in early trials in ovarian and renal cancers [296,297,298]. Similarly, tocilizumab (MRA) is a humanized antihuman IL-6R antibody that inhibits IL-6 signaling [299]. Based on the importance of IL-6 in regulating NASH and HCC, in principle, the IL-6 blockade might be beneficial in obesity-induced liver cancer. However, as of now, this strategy has not been reported.
A summary of the various avenues as effective therapeutic regimens targeting adipokines is listed in Table 3.
Table 3.
Adiponectin and adipokines as therapeutic targets in cancer.
Table 3.
Adiponectin and adipokines as therapeutic targets in cancer.
| Inhibitors | Mechanism | Therapeutic Effect | References |
|---|---|---|---|
| ADP-355 | Adiponectin receptor agonist | Suppress tumor growth by binding to AdipoR1 and AdipoR2 | [268] |
| Efatutazone | PPARγ agonist | Delay invasive progression in mammary DCIS and induce differentiation of cancer | [275] |
| Rosiglitazone | ADPN expression enhancer Leptin signaling inhibitor | Suppress growth, migration, and invasion of cancer cells by inhibiting | [277] |
| Troglitazone | PPARγ activator | Suppresses growth, migration, and invasion of cancer cells | [276,277] |
| PEG-LPrA2 | Interferes with the proangiogenic factors and proproliferative factors | Inhibit tumor growth | [283] |
| LDFI | Leptin peptide antagonist binding to the leptin binding site I | Suppresses the growth of cancer cells | [285] |
| AG490 | Suppress leptin/Jak2/STAT3 signaling | Inhibits cell proliferation | [286,287,288] |
| AM063 and AM095 | ATX inhibitor and LPAR1 antagonist, respectively | Protects from hepatic fibrosis and abrogates HCC development | [292,293] |
| CNTO 328 (siltuximab) | IL-6/IL-6R/gp130 trans-signaling inhibitor | Suppress tumor growth by interfering in the binding of IL-6 and IL-6R | [296,297] |
| Triacsin C | TAG accumulation into lipid droplets | Inhibits apoptosis by targeting intracellular long-chain acyl-CoA synthetases in NASH | [300] |
| Isoquercitrin | Activates glucagon-like peptide-1 | Targets dipeptidyl peptidase-IV and inhibits apoptotic signaling in NASH | [301] |
7. Conclusions
A multitude of studies place AT as a pivotal regulator of obesity-associated metabolic disorders, such as IR, dyslipidemia, and NAFLD. Enlarged AT leads to macrophage infiltration, and an imbalance between proinflammatory and anti-inflammatory factors secreted by AT leads to inflammation, impaired insulin sensitivity, and the dysregulation of lipid metabolism. Excessive FFA released by AT aids in the initiation and progression of obesity-induced metabolic complications, inducing steatosis and steatohepatitis in the liver, ultimately leading to the development of liver cancer. An unfavorable adipokine profile, induced by obesity, with reduced adipokines having anti-inflammatory or antitumor activity, further paves the way for hepatocarcinogenesis. An imbalanced calorie intake and high insulin levels stimulate lipid accumulation in adipocytes, thus suppressing ADP secretion, leading to an inflammatory state by macrophage infiltration (Figure 2). The subsequent lipolysis leads to the accumulation of triglycerides in visceral adipose tissue (VAT) through secreted FAs, predisposing to steatosis, NASH to HCC. Obese AT promotes the inflammatory response, thus damaging hepatic cells and impairing immune response. These observations open up novel avenues for therapeutic intervention of obesity-associated hepatic malignancies. It will be worthwhile to perform an in-depth exploration of the role of existing ADP agonists, AT, and apoptosis inhibitors administered in other cancers (Table 3) for regulating adipokine-mediated obesity-induced HCC. The correlation between adipose tissue and adipokines with the development of obesity-induced HCC and the probable impact of adiponectin agonists and other inhibitors are summarized in Figure 2.
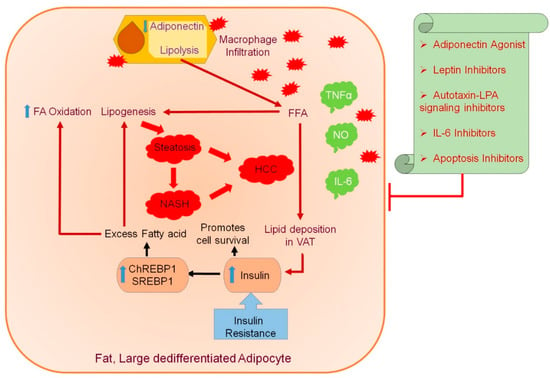
Figure 2.
Correlation between adipose tissue and adipokines with the development of obesity-induced hepatocellular carcinoma (HCC) and the effect of adiponectin agonists and other inhibitors. The adipose tissue (AT) secretes various bioactive peptides, known as adipokines, and their associated dysregulation has been implicated in metabolic disorders caused by obesity. The swollen and dedifferentiated adipocytes secrete less adiponectin (ADP) during obesity, and ADP is the only adipokine of which the circulating levels are significantly reduced during obesity. In muscles and the liver, ADP increases insulin action and exerts an antiatherogenic effect. Leptin is another adipokine that regulates energy balance and exerts an insulin-sensitizing effect, but its beneficial effects are reduced in obesity due to leptin resistance. In obese individuals, an inflammatory state is contributed through macrophage infiltration due to excessive production of TNFα, IL-6, or resistin deteriorating insulin action in the liver. Increased lipolysis secretes FAs that accumulate triglyceride in visceral adipose tissue (VAT) in correlation with insulin resistance (IR). The IR and elevated FFAs predispose to steatosis, promoting hepatic lipogenesis and increasing SREBP1 and ChREBP1 expression, further promoting damage to liver tissues and a state of inflammatory response, resulting in NASH followed by HCC. The inflammatory response promoted by obese AT contributes to damage of hepatic cells and impaired immune response. The administration of ADP agonists and AT targeting inhibitors might be a promising therapeutic target for adipokine-mediated obesity-induced HCC. Blue upward arrow indicates an increase and blue downward arrow indicates a decrease.
In summary, the humongous metabolic catastrophe, associated with obesity, creates a major hindrance in the effective implementation of anticancer strategies that have been shown to be effective in cancers in nonobese individuals. In-depth future research will further illuminate the functions and mechanisms of AT-secreted biomolecules for a better understanding of the underlying molecular mechanism of obesity-associated liver cancer. This knowledge will help design appropriate combinatorial therapy to target both the metabolic abnormalities and eliminate malignant cells to effectively combat the disease. The identification of molecules that can regulate both metabolic and oncogenic abnormalities will also help develop effective, targeted therapy. Our studies over the years have documented a critical role of the oncogene astrocyte elevated gene-1 (AEG-1) in regulating both steatosis and inflammation, activating PI3K/Akt signaling and contributing to IR, as well as driving the development and progression of HCC [302,303,304,305]. We documented that the delivery of AEG-1 siRNA using a hepatocyte-targeted nanoparticle could effectively protect from HFD-induced NASH and inhibit orthotopic human HCC xenografts [302,305]. Further comprehensive studies are required to determine the efficacy of this strategy in a model of NASH-HCC and interrogate the role of AEG-1 and similar molecules in regulating AT function.
Funding
The present study was supported in part by The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1R01DK107451-01A1, National Cancer Institute (NCI) Grants 1R01CA230561-01A1, 1R01CA240004-01, and 1R01CA244993-01, and Department of Defense (DOD) Grant CA170048.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rajesh, Y.; Sarkar, D. Molecular Mechanisms Regulating Obesity-Associated Hepatocellular Carcinoma. Cancers 2020, 12, 1290. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Ronti, T.; Lupattelli, G.; Mannarino, E. The endocrine function of adipose tissue: An update. Clin. Endocrinol. 2006, 64, 355–365. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Poulos, S.P.; Hausman, D.B.; Hausman, G.J. The development and endocrine functions of adipose tissue. Mol. Cell. Endocrinol. 2010, 323, 20–34. [Google Scholar] [CrossRef]
- Proenca, A.R.; Sertie, R.A.; Oliveira, A.C.; Campana, A.B.; Caminhotto, R.O.; Chimin, P.; Lima, F.B. New concepts in white adipose tissue physiology. Braz. J. Med. Biol. Res. 2014, 47, 192–205. [Google Scholar] [CrossRef]
- Vazquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef]
- Esteve Rafols, M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinol. Nutr. 2014, 61, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.T.; Hochfeld, W.E.; Myburgh, R.; Pepper, M.S. Adipocyte and adipogenesis. Eur. J. Cell Biol. 2013, 92, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Scarpato, R.; Verola, C.; Fabiani, B.; Bianchi, V.; Saggese, G.; Federico, G. Nuclear damage in peripheral lymphocytes of obese and overweight Italian children as evaluated by the gamma-H2AX focus assay and micronucleus test. FASEB J. 2011, 25, 685–693. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef]
- Adams, J.P.; Murphy, P.G. Obesity in anaesthesia and intensive care. Br. J. Anaesth. 2000, 85, 91–108. [Google Scholar] [CrossRef]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Johnson, C.L. Prevalence and trends in obesity among US adults, 1999–2000. JAMA 2002, 288, 1723–1727. [Google Scholar] [CrossRef]
- Hedley, A.A.; Ogden, C.L.; Johnson, C.L.; Carroll, M.D.; Curtin, L.R.; Flegal, K.M. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA 2004, 291, 2847–2850. [Google Scholar] [CrossRef]
- Chow, W.H.; Gridley, G.; Fraumeni, J.F., Jr.; Jarvholm, B. Obesity, hypertension, and the risk of kidney cancer in men. N. Engl. J. Med. 2000, 343, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.C.; Thomas, C.; Bowman, B.A. Obesity and endometrial cancer: Challenges for public health action. Womens Health 2009, 5, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Majed, B.; Moreau, T.; Senouci, K.; Salmon, R.J.; Fourquet, A.; Asselain, B. Is obesity an independent prognosis factor in woman breast cancer? Breast Cancer Res. Treat. 2008, 111, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Schapira, D.V.; Kumar, N.B.; Lyman, G.H.; Cox, C.E. Abdominal obesity and breast cancer risk. Ann. Intern. Med. 1990, 112, 182–186. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Yu, M.C.; Yuan, J.M. Environmental factors and risk for hepatocellular carcinoma. Gastroenterology 2004, 127, S72–S78. [Google Scholar] [CrossRef]
- Chen, C.L.; Yang, H.I.; Yang, W.S.; Liu, C.J.; Chen, P.J.; You, S.L.; Wang, L.Y.; Sun, C.A.; Lu, S.N.; Chen, D.S.; et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: A follow-up study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef]
- Diehl, A.M. Hepatic complications of obesity. Gastroenterol. Clin. N. Am. 2010, 39, 57–68. [Google Scholar] [CrossRef]
- Tarantino, G.; Colicchio, P.; Conca, P.; Finelli, C.; Di Minno, M.N.; Tarantino, M.; Capone, D.; Pasanisi, F. Young adult obese subjects with and without insulin resistance: What is the role of chronic inflammation and how to weigh it non-invasively? J. Inflamm. 2009, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- de Alwis, N.M.; Day, C.P. Non-alcoholic fatty liver disease: The mist gradually clears. J. Hepatol. 2008, 48 (Suppl. S1), S104–S112. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef]
- Renquist, B.J.; Murphy, J.G.; Larson, E.A.; Olsen, D.; Klein, R.F.; Ellacott, K.L.; Cone, R.D. Melanocortin-3 receptor regulates the normal fasting response. Proc. Natl. Acad. Sci. USA 2012, 109, E1489–E1498. [Google Scholar] [CrossRef]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef]
- Awazawa, M.; Ueki, K.; Inabe, K.; Yamauchi, T.; Kaneko, K.; Okazaki, Y.; Bardeesy, N.; Ohnishi, S.; Nagai, R.; Kadowaki, T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem. Biophys. Res. Commun. 2009, 382, 51–56. [Google Scholar] [CrossRef]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Matsuzaka, T.; Ide, T.; Yoshikawa, T.; Amemiya-Kudo, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J. Biol. Chem. 2002, 277, 19353–19357. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Larsen, P. FGF21 reloaded: Challenges of a rapidly growing field. Trends Endocrinol. Metab. 2011, 22, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Yang, G.; Yu, X.; Soukas, A.A.; Wolfish, C.S.; Friedman, J.M.; Li, C. Induction of leptin receptor expression in the liver by leptin and food deprivation. J. Biol. Chem. 2005, 280, 10034–10039. [Google Scholar] [CrossRef]
- Nishina, P.M.; Lowe, S.; Wang, J.; Paigen, B. Characterization of plasma lipids in genetically obese mice: The mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism 1994, 43, 549–553. [Google Scholar] [CrossRef]
- Haluzik, M.; Colombo, C.; Gavrilova, O.; Chua, S.; Wolf, N.; Chen, M.; Stannard, B.; Dietz, K.R.; Le Roith, D.; Reitman, M.L. Genetic background (C57BL/6J versus FVB/N) strongly influences the severity of diabetes and insulin resistance in ob/ob mice. Endocrinology 2004, 145, 3258–3264. [Google Scholar] [CrossRef]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef]
- Huynh, F.K.; Neumann, U.H.; Wang, Y.; Rodrigues, B.; Kieffer, T.J.; Covey, S.D. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology 2013, 57, 543–554. [Google Scholar] [CrossRef]
- Huang, W.; Dedousis, N.; Bandi, A.; Lopaschuk, G.D.; O’Doherty, R.M. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology 2006, 147, 1480–1487. [Google Scholar] [CrossRef]
- Kamada, Y.; Tamura, S.; Kiso, S.; Matsumoto, H.; Saji, Y.; Yoshida, Y.; Fukui, K.; Maeda, N.; Nishizawa, H.; Nagaretani, H.; et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 2003, 125, 1796–1807. [Google Scholar] [CrossRef]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef]
- Javor, E.D.; Ghany, M.G.; Cochran, E.K.; Oral, E.A.; DePaoli, A.M.; Premkumar, A.; Kleiner, D.E.; Gorden, P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005, 41, 753–760. [Google Scholar] [CrossRef]
- Hausman, D.B.; DiGirolamo, M.; Bartness, T.J.; Hausman, G.J.; Martin, R.J. The biology of white adipocyte proliferation. Obes. Rev. 2001, 2, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef]
- Weyer, C.; Foley, J.E.; Bogardus, C.; Tataranni, P.A.; Pratley, R.E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mokan, M.; Simoneau, J.A.; Mandarino, L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Investig. 1993, 92, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef]
- Jernas, M.; Palming, J.; Sjoholm, K.; Jennische, E.; Svensson, P.A.; Gabrielsson, B.G.; Levin, M.; Sjogren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FASEB J. 2006, 20, 1540–1542. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Amrani, A.; Jafarian-Tehrani, M.; Mormede, P.; Durant, S.; Pleau, J.M.; Haour, F.; Dardenne, M.; Homo-Delarche, F. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J. Endocrinol. 1996, 148, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Chen, Q.; Shah, S.; Du, J.; Tao, B.; Tzameli, I.; Yan, W.; Xu, H. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: Involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 2009, 58, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Thacker, R.I.; Hall, B.E.; Kong, R.; Granneman, J.G. Exploring the activated adipogenic niche: Interactions of macrophages and adipocyte progenitors. Cell Cycle 2014, 13, 184–190. [Google Scholar] [CrossRef]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschop, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Nara, N.; Nakayama, Y.; Okamoto, S.; Tamura, H.; Kiyono, M.; Muraoka, M.; Tanaka, K.; Taya, C.; Shitara, H.; Ishii, R.; et al. Disruption of CXC motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J. Biol. Chem. 2007, 282, 30794–30803. [Google Scholar] [CrossRef]
- Clement, K.; Viguerie, N.; Poitou, C.; Carette, C.; Pelloux, V.; Curat, C.A.; Sicard, A.; Rome, S.; Benis, A.; Zucker, J.D.; et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004, 18, 1657–1669. [Google Scholar] [CrossRef]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab. 2012, 23, 407–415. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- de Heredia, F.P.; Gomez-Martinez, S.; Marcos, A. Obesity, inflammation and the immune system. Proc. Nutr. Soc. 2012, 71, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Hebbard, L.; Ranscht, B. Multifaceted roles of adiponectin in cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 59–69. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Smitka, K.; Maresova, D. Adipose Tissue as an Endocrine Organ: An Update on Pro-inflammatory and Anti-inflammatory Microenvironment. Prague Med. Rep. 2015, 116, 87–111. [Google Scholar] [CrossRef]
- Hursting, S.D.; Dunlap, S.M. Obesity, metabolic dysregulation, and cancer: A growing concern and an inflammatory (and microenvironmental) issue. Ann. N. Y. Acad. Sci. 2012, 1271, 82–87. [Google Scholar] [CrossRef]
- de Ferranti, S.; Mozaffarian, D. The perfect storm: Obesity, adipocyte dysfunction, and metabolic consequences. Clin. Chem. 2008, 54, 945–955. [Google Scholar] [CrossRef]
- Sommer, G.; Kralisch, S.; Stangl, V.; Vietzke, A.; Kohler, U.; Stepan, H.; Faber, R.; Schubert, A.; Lossner, U.; Bluher, M.; et al. Secretory products from human adipocytes stimulate proinflammatory cytokine secretion from human endothelial cells. J. Cell. Biochem. 2009, 106, 729–737. [Google Scholar] [CrossRef]
- Fasshauer, M.; Bluher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Clinical relevance of adipokines. Diabetes Metab. J. 2012, 36, 317–327. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; de Courten, M.P.; Stojanovska, L.; Blatch, G.L.; Tangalakis, K.; de Courten, B. The complex immunological and inflammatory network of adipose tissue in obesity. Mol. Nutr. Food Res. 2016, 60, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J. Adipokines influence the inflammatory balance in autoimmunity. Cytokine 2015, 75, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.E.; Marzullo, P.; Ricotti, R.; Bona, G.; Prodam, F. The pathophysiology of abdominal adipose tissue depots in health and disease. Horm. Mol. Biol. Clin. Investig. 2014, 19, 57–74. [Google Scholar] [CrossRef]
- Badoud, F.; Perreault, M.; Zulyniak, M.A.; Mutch, D.M. Molecular insights into the role of white adipose tissue in metabolically unhealthy normal weight and metabolically healthy obese individuals. FASEB J. 2015, 29, 748–758. [Google Scholar] [CrossRef]
- Ghigliotti, G.; Barisione, C.; Garibaldi, S.; Fabbi, P.; Brunelli, C.; Spallarossa, P.; Altieri, P.; Rosa, G.; Spinella, G.; Palombo, D.; et al. Adipose tissue immune response: Novel triggers and consequences for chronic inflammatory conditions. Inflammation 2014, 37, 1337–1353. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, P.; Sahakyan, K.R.; Davison, D.E.; Sert-Kuniyoshi, F.H.; Romero-Corral, A.; Swain, J.M.; Jensen, M.D.; Lopez-Jimenez, F.; Kara, T.; et al. Differential effects of leptin on adiponectin expression with weight gain versus obesity. Int. J. Obes. 2016, 40, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Jovinge, S.; Hamsten, A.; Tornvall, P.; Proudler, A.; Bavenholm, P.; Ericsson, C.G.; Godsland, I.; de Faire, U.; Nilsson, J. Evidence for a role of tumor necrosis factor alpha in disturbances of triglyceride and glucose metabolism predisposing to coronary heart disease. Metabolism 1998, 47, 113–118. [Google Scholar] [CrossRef]
- Feingold, K.R.; Serio, M.K.; Adi, S.; Moser, A.H.; Grunfeld, C. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion. Endocrinology 1989, 124, 2336–2342. [Google Scholar] [CrossRef]
- Kawakami, M.; Cerami, A. Studies of endotoxin-induced decrease in lipoprotein lipase activity. J. Exp. Med. 1981, 154, 631–639. [Google Scholar] [CrossRef]
- Qin, B.; Anderson, R.A.; Adeli, K. Tumor necrosis factor-alpha directly stimulates the overproduction of hepatic apolipoprotein B100-containing VLDL via impairment of hepatic insulin signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1120–G1129. [Google Scholar] [CrossRef]
- Mohrschladt, M.F.; Weverling-Rijnsburger, A.W.; de Man, F.H.; Stoeken, D.J.; Sturk, A.; Smelt, A.H.; Westendorp, R.G. Hyperlipoproteinemia affects cytokine production in whole blood samples ex vivo. The influence of lipid-lowering therapy. Atherosclerosis 2000, 148, 413–419. [Google Scholar] [CrossRef]
- Jonkers, I.J.; Mohrschladt, M.F.; Westendorp, R.G.; van der Laarse, A.; Smelt, A.H. Severe hypertriglyceridemia with insulin resistance is associated with systemic inflammation: Reversal with bezafibrate therapy in a randomized controlled trial. Am. J. Med. 2002, 112, 275–280. [Google Scholar] [CrossRef]
- Van Exel, E.; Gussekloo, J.; de Craen, A.J.; Frolich, M.; Bootsma-Van Der Wiel, A.; Westendorp, R.G. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: The Leiden 85-Plus Study. Diabetes 2002, 51, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Grunfeld, C.; Dinarello, C.A.; Feingold, K.R. Tumor necrosis factor-alpha, interleukin-1, and interferon alpha stimulate triglyceride synthesis in HepG2 cells. Metabolism 1991, 40, 894–898. [Google Scholar] [CrossRef]
- Yang, Y.; Ju, D.; Zhang, M.; Yang, G. Interleukin-6 stimulates lipolysis in porcine adipocytes. Endocrine 2008, 33, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Hardardottir, I.; Doerrler, W.; Feingold, K.R.; Grunfeld, C. Cytokines stimulate lipolysis and decrease lipoprotein lipase activity in cultured fat cells by a prostaglandin independent mechanism. Biochem. Biophys. Res. Commun. 1992, 186, 237–243. [Google Scholar] [CrossRef]
- Hardardottir, I.; Moser, A.H.; Memon, R.; Grunfeld, C.; Feingold, K.R. Effects of TNF, IL-1, and the combination of both cytokines on cholesterol metabolism in Syrian hamsters. Lymphokine Cytokine Res. 1994, 13, 161–166. [Google Scholar]
- Kawakami, M.; Murase, T.; Ogawa, H.; Ishibashi, S.; Mori, N.; Takaku, F.; Shibata, S. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J. Biochem. 1987, 101, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Nordan, R.P.; McIntosh, J.; Calvo, J.C.; Scow, R.O.; Jablons, D. Interleukin 6 reduces lipoprotein lipase activity in adipose tissue of mice in vivo and in 3T3-L1 adipocytes: A possible role for interleukin 6 in cancer cachexia. Cancer Res. 1992, 52, 4113–4116. [Google Scholar]
- Fernandez-Real, J.M.; Gutierrez, C.; Ricart, W.; Castineira, M.J.; Vendrell, J.; Richart, C. Plasma levels of the soluble fraction of tumor necrosis factor receptors 1 and 2 are independent determinants of plasma cholesterol and LDL-cholesterol concentrations in healthy subjects. Atherosclerosis 1999, 146, 321–327. [Google Scholar] [CrossRef]
- Skoog, T.; Dichtl, W.; Boquist, S.; Skoglund-Andersson, C.; Karpe, F.; Tang, R.; Bond, M.G.; de Faire, U.; Nilsson, J.; Eriksson, P.; et al. Plasma tumour necrosis factor-alpha and early carotid atherosclerosis in healthy middle-aged men. Eur. Heart J. 2002, 23, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, W.H.; Miller, L.D.; Albers, J.J.; Smith, T.K.; Parks, J.S. Lipopolysaccharide and tumor necrosis factor cause a fall in plasma concentration of lecithin: Cholesterol acyltransferase in cynomolgus monkeys. J. Lipid Res. 1990, 31, 1099–1107. [Google Scholar] [CrossRef]
- Ruan, X.Z.; Varghese, Z.; Fernando, R.; Moorhead, J.F. Cytokine regulation of low-density lipoprotein receptor gene transcription in human mesangial cells. Nephrol. Dial. Transplant. 1998, 13, 1391–1397. [Google Scholar] [CrossRef]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Dysregulation of LDL receptor under the influence of inflammatory cytokines: A new pathway for foam cell formation. Kidney Int. 2001, 60, 1716–1725. [Google Scholar] [CrossRef]
- Ruan, X.Z.; Moorhead, J.F.; Fernando, R.; Wheeler, D.C.; Powis, S.H.; Varghese, Z. Regulation of lipoprotein trafficking in the kidney: Role of inflammatory mediators and transcription factors. Biochem. Soc. Trans. 2004, 32, 88–91. [Google Scholar] [CrossRef]
- Bartolome, N.; Rodriguez, L.; Martinez, M.J.; Ochoa, B.; Chico, Y. Upregulation of apolipoprotein B secretion, but not lipid, by tumor necrosis factor-alpha in rat hepatocyte cultures in the absence of extracellular fatty acids. Ann. N. Y. Acad. Sci. 2007, 1096, 55–69. [Google Scholar] [CrossRef]
- Crowl, R.M.; Stoller, T.J.; Conroy, R.R.; Stoner, C.R. Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. J. Biol. Chem. 1991, 266, 2647–2651. [Google Scholar] [CrossRef]
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C.S. Adiponectin and cancer: A systematic review. Br. J. Cancer 2006, 94, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Beales, I.L. Adiponectin stimulates proliferation and cytokine secretion in colonic epithelial cells. Regul. Pept. 2006, 134, 105–113. [Google Scholar] [CrossRef]
- Lin, H.; Yu, C.H.; Jen, C.Y.; Cheng, C.F.; Chou, Y.; Chang, C.C.; Juan, S.H. Adiponectin-mediated heme oxygenase-1 induction protects against iron-induced liver injury via a PPARalpha dependent mechanism. Am. J. Pathol. 2010, 177, 1697–1709. [Google Scholar] [CrossRef]
- Nepal, S.; Kim, M.J.; Subedi, A.; Lee, E.S.; Yong, C.S.; Kim, J.A.; Kang, W.; Kwak, M.K.; Arya, D.S.; Park, P.H. Globular adiponectin inhibits ethanol-induced apoptosis in HepG2 cells through heme oxygenase-1 induction. Biochem. Pharmacol. 2012, 84, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, I.; Bechmann, L.P.; Odenthal, M.; Jochum, C.; Marquitan, G.; Drebber, U.; Gerken, G.; Gieseler, R.K.; Dienes, H.P.; Canbay, A. Adiponectin inhibits steatotic CD95/Fas up-regulation by hepatocytes: Therapeutic implications for hepatitis C. J. Hepatol. 2009, 50, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liao, Y.; Li, Q.; Chen, M.; Zhao, Q.; Deng, R.; Wu, C.; Yang, A.; Guo, Z.; Wang, D.; et al. Recombinant adiponectin ameliorates liver ischemia reperfusion injury via activating the AMPK/eNOS pathway. PLoS ONE 2013, 8, e66382. [Google Scholar] [CrossRef]
- Wanninger, J.; Bauer, S.; Eisinger, K.; Weiss, T.S.; Walter, R.; Hellerbrand, C.; Schaffler, A.; Higuchi, A.; Walsh, K.; Buechler, C. Adiponectin upregulates hepatocyte CMKLR1 which is reduced in human fatty liver. Mol. Cell. Endocrinol. 2012, 349, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, J.; Neumeier, M.; Weigert, J.; Bauer, S.; Weiss, T.S.; Schaffler, A.; Krempl, C.; Bleyl, C.; Aslanidis, C.; Scholmerich, J.; et al. Adiponectin-stimulated CXCL8 release in primary human hepatocytes is regulated by ERK1/ERK2, p38 MAPK, NF-kappaB, and STAT3 signaling pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G611–G618. [Google Scholar] [CrossRef]
- Court, F.G.; Wemyss-Holden, S.A.; Dennison, A.R.; Maddern, G.J. The mystery of liver regeneration. Br. J. Surg. 2002, 89, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Parsons, C.J.; Stefanovic, B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 2006, 45, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Pratt, B.T.; Barnes, M.; McMullen, M.R.; Nagy, L.E. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: Link between the metabolic and innate immune activity of full-length adiponectin. J. Biol. Chem. 2011, 286, 13460–13469. [Google Scholar] [CrossRef] [PubMed]
- Akifusa, S.; Kamio, N.; Shimazaki, Y.; Yamaguchi, N.; Nishihara, T.; Yamashita, Y. Globular adiponectin-induced RAW 264 apoptosis is regulated by a reactive oxygen species-dependent pathway involving Bcl-2. Free Radic. Biol. Med. 2009, 46, 1308–1316. [Google Scholar] [CrossRef]
- Akifusa, S.; Kamio, N.; Shimazaki, Y.; Yamaguchi, N.; Yamashita, Y. Regulation of globular adiponectin-induced apoptosis by reactive oxygen/nitrogen species in RAW264 macrophages. Free Radic. Biol. Med. 2008, 45, 1326–1339. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Xu, A.; Srinivasan, S.; Anania, F.A. The roles of leptin and adiponectin: A novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am. J. Pathol. 2005, 166, 1655–1669. [Google Scholar] [CrossRef]
- Qamar, A.; Sheikh, S.Z.; Masud, A.; Jhandier, M.N.; Inayat, I.B.; Hakim, W.; Mehal, W.Z. In vitro and in vivo protection of stellate cells from apoptosis by leptin. Dig. Dis. Sci. 2006, 51, 1697–1705. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Schroyen, B.; Guimaraes, E.L.; Dolle, L.; Coulon, S.; Empsen, C.; Nyssen, M.; Geerts, A.; Colle, I.; Geerts, A.; van Grunsven, L.A. Leptin-mediated reactive oxygen species production does not significantly affect primary mouse hepatocyte functions in vitro. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1370–1380. [Google Scholar] [CrossRef]
- Mistry, T.; Digby, J.E.; Desai, K.M.; Randeva, H.S. Leptin and adiponectin interact in the regulation of prostate cancer cell growth via modulation of p53 and bcl-2 expression. BJU Int. 2008, 101, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lei, W.; Shen, L.; Luo, H.S.; Shen, Z.X. Primary study of leptin and human hepatocellular carcinoma in vitro. World J. Gastroenterol. 2008, 14, 2900–2904. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, J.; Liu, M.; An, M.; Lei, L.; Guo, W. Human leptin protein activates the growth of HepG2 cells by inhibiting PERKmediated ER stress and apoptosis. Mol. Med. Rep. 2014, 10, 1649–1655. [Google Scholar] [CrossRef]
- Ramani, K.; Yang, H.; Xia, M.; Ara, A.I.; Mato, J.M.; Lu, S.C. Leptin’s mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology 2008, 47, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Nepal, S.; Kim, M.J.; Hong, J.T.; Kim, S.H.; Sohn, D.H.; Lee, S.H.; Song, K.; Choi, D.Y.; Lee, E.S.; Park, P.H. Autophagy induction by leptin contributes to suppression of apoptosis in cancer cells and xenograft model: Involvement of p53/FoxO3A axis. Oncotarget 2015, 6, 7166–7181. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Akazawa, Y.; Cazanave, S.C.; Bronk, S.F.; Elmi, N.A.; Werneburg, N.W.; Billadeau, D.D.; Gores, G.J. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J. Hepatol. 2011, 54, 765–772. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Gores, G.J.; Hirsova, P.; Kirby, M.; Miles, L.; Jaeschke, A.; Kohli, R. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver Int. 2014, 34, 427–437. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Hsieh, S.; Leaderer, B.P.; Feldstein, A.E.; Santoro, N.; McKay, L.A.; Caprio, S.; McConnell, R. Traffic-related air pollution associations with cytokeratin-18, a marker of hepatocellular apoptosis, in an overweight and obese paediatric population. Pediatr. Obes. 2018, 13, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, K.; Cazanave, S.C.; Werneburg, N.W.; Razumilava, N.; Mertens, J.C.; Bronk, S.F.; Gores, G.J. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J. Hepatol. 2012, 57, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.S.; Lindblom, K.R.; Robeson, A.; Stevens, R.D.; Ilkayeva, O.R.; Newgard, C.B.; Kornbluth, S.; Andersen, J.L. Metabolomic profiling reveals a role for caspase-2 in lipoapoptosis. J. Biol. Chem. 2013, 288, 14463–14475. [Google Scholar] [CrossRef]
- Li, C.P.; Li, J.H.; He, S.Y.; Li, P.; Zhong, X.L. Roles of Fas/Fasl, Bcl-2/Bax, and Caspase-8 in rat nonalcoholic fatty liver disease pathogenesis. Genet. Mol. Res. 2014, 13, 3991–3999. [Google Scholar] [CrossRef]
- El Bassat, H.; Ziada, D.H.; Hasby, E.A.; Nagy, H.; Abo Ryia, M.H. Apoptotic and anti-apoptotic seromarkers for assessment of disease severity of non-alcoholic steatohepatitis. Arab. J. Gastroenterol. 2014, 15, 6–11. [Google Scholar] [CrossRef]
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Physiol. 2012, 227, 1130–1137. [Google Scholar] [CrossRef]
- Eguchi, A.; De Mollerat Du Jeu, X.; Johnson, C.D.; Nektaria, A.; Feldstein, A.E. Liver Bid suppression for treatment of fibrosis associated with non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Zhao, L.P.; Tian, X.X.; Yan, C.H.; Li, Y.; Liu, Y.X.; Wang, P.X.; Zhang, X.J.; Han, Y.L. The novel intracellular protein CREG inhibits hepatic steatosis, obesity, and insulin resistance. Hepatology 2017, 66, 834–854. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.L.; de Oliveira Matoso, R.; Teixeira, L.G.; Menezes, Z.; Pereira, S.S.; Alves, A.C.; Batista, N.V.; de Faria, A.M.; Cara, D.C.; Ferreira, A.V.; et al. Gluten-free diet reduces adiposity, inflammation and insulin resistance associated with the induction of PPAR-alpha and PPAR-gamma expression. J. Nutr. Biochem. 2013, 24, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Ozcan, L.; Ghorpade, D.S.; Ferrante, A.W., Jr.; Tabas, I. Suppression of Adaptive Immune Cell Activation Does Not. Alter Innate Immune Adipose Inflammation or Insulin Resistance in Obesity. PLoS ONE 2015, 10, e0135842. [Google Scholar]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef]
- Wieser, V.; Moschen, A.R.; Tilg, H. Inflammation, cytokines and insulin resistance: A clinical perspective. Arch. Immunol. Ther. Exp. 2013, 61, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gill, R.; Pedersen, T.L.; Higgins, L.J.; Newman, J.W.; Rutledge, J.C. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J. Lipid Res. 2009, 50, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Kanety, H.; Feinstein, R.; Papa, M.Z.; Hemi, R.; Karasik, A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J. Biol. Chem. 1995, 270, 23780–23784. [Google Scholar] [CrossRef] [PubMed]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Mooney, R.A. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002, 51, 3391–3399. [Google Scholar] [CrossRef]
- He, J.; Usui, I.; Ishizuka, K.; Kanatani, Y.; Hiratani, K.; Iwata, M.; Bukhari, A.; Haruta, T.; Sasaoka, T.; Kobayashi, M. Interleukin-1alpha inhibits insulin signaling with phosphorylating insulin receptor substrate-1 on serine residues in 3T3-L1 adipocytes. Mol. Endocrinol. 2006, 20, 114–124. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Meijer, K.; de Vries, M.; Al-Lahham, S.; Bruinenberg, M.; Weening, D.; Dijkstra, M.; Kloosterhuis, N.; van der Leij, R.J.; van der Want, H.; Kroesen, B.J.; et al. Human primary adipocytes exhibit immune cell function: Adipocytes prime inflammation independent of macrophages. PLoS ONE 2011, 6, e17154. [Google Scholar] [CrossRef]
- Keophiphath, M.; Rouault, C.; Divoux, A.; Clement, K.; Lacasa, D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Lazennec, G.; Lagarrigue, S.; Clape, C.; Iankova, I.; Teyssier, J.; Annicotte, J.S.; Schmidt, J.; Mataki, C.; Yamamoto, H.; et al. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab. 2009, 9, 339–349. [Google Scholar] [CrossRef]
- Ruan, H.; Lodish, H.F. Insulin resistance in adipose tissue: Direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003, 14, 447–455. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Pipek, R.; Mandarino, L.J.; DeFronzo, R.A. Tumor necrosis factor alpha and insulin resistance in obese type 2 diabetic patients. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 88–94. [Google Scholar] [CrossRef]
- Wascher, T.C.; Lindeman, J.H.; Sourij, H.; Kooistra, T.; Pacini, G.; Roden, M. Chronic TNF-alpha neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol. Med. 2011, 17, 189–193. [Google Scholar] [CrossRef]
- Bernstein, L.E.; Berry, J.; Kim, S.; Canavan, B.; Grinspoon, S.K. Effects of etanercept in patients with the metabolic syndrome. Arch. Intern. Med. 2006, 166, 902–908. [Google Scholar] [CrossRef] [PubMed]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef]
- Fernandez-Real, J.M.; Ricart, W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr. Rev. 2003, 24, 278–301. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [PubMed]
- Sopasakis, V.R.; Sandqvist, M.; Gustafson, B.; Hammarstedt, A.; Schmelz, M.; Yang, X.; Jansson, P.A.; Smith, U. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obes. Res. 2004, 12, 454–460. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Van Nhieu, J.T.; Jardel, C.; Bruckert, E.; Grimaldi, A.; Robert, J.J.; Capeau, J.; Hainque, B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2084–2089. [Google Scholar] [CrossRef]
- Wood, I.S.; Wang, B.; Jenkins, J.R.; Trayhurn, P. The pro-inflammatory cytokine IL-18 is expressed in human adipose tissue and strongly upregulated by TNFalpha in human adipocytes. Biochem. Biophys. Res. Commun. 2005, 337, 422–429. [Google Scholar] [CrossRef]
- Tan, H.W.; Liu, X.; Bi, X.P.; Xing, S.S.; Li, L.; Gong, H.P.; Zhong, M.; Wang, Z.H.; Zhang, Y.; Zhang, W. IL-18 overexpression promotes vascular inflammation and remodeling in a rat model of metabolic syndrome. Atherosclerosis 2010, 208, 350–357. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Leptin, leptin receptors and the control of body weight. Eur. J. Med. Res. 1997, 2, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Mozzoli, M.; Ryan, I. Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 1996, 81, 3419–3423. [Google Scholar]
- Sarraf, P.; Frederich, R.C.; Turner, E.M.; Ma, G.; Jaskowiak, N.T.; Rivet, D.J., 3rd; Flier, J.S.; Lowell, B.B.; Fraker, D.L.; Alexander, H.R. Multiple cytokines and acute inflammation raise mouse leptin levels: Potential role in inflammatory anorexia. J. Exp. Med. 1997, 185, 171–175. [Google Scholar] [CrossRef]
- Grunfeld, C.; Zhao, C.; Fuller, J.; Pollack, A.; Moser, A.; Friedman, J.; Feingold, K.R. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J. Clin. Investig. 1996, 97, 2152–2157. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.; Franco, C.B.; Wang, K.B.; Wong, M.L.; Licinio, J. Leptin: Molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq. Bras. Endocrinol. Metabol. 2012, 56, 597–607. [Google Scholar] [CrossRef]
- Marroqui, L.; Gonzalez, A.; Neco, P.; Caballero-Garrido, E.; Vieira, E.; Ripoll, C.; Nadal, A.; Quesada, I. Role of leptin in the pancreatic beta-cell: Effects and signaling pathways. J. Mol. Endocrinol. 2012, 49, R9–R17. [Google Scholar] [CrossRef]
- Perez, C.; Fernandez-Galaz, C.; Fernandez-Agullo, T.; Arribas, C.; Andres, A.; Ros, M.; Carrascosa, J.M. Leptin impairs insulin signaling in rat adipocytes. Diabetes 2004, 53, 347–353. [Google Scholar] [CrossRef]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Steppan, C.M.; Lazar, M.A. The current biology of resistin. J. Intern. Med. 2004, 255, 439–447. [Google Scholar] [CrossRef]
- Banerjee, R.R.; Rangwala, S.M.; Shapiro, J.S.; Rich, A.S.; Rhoades, B.; Qi, Y.; Wang, J.; Rajala, M.W.; Pocai, A.; Scherer, P.E.; et al. Regulation of fasted blood glucose by resistin. Science 2004, 303, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Nie, Z.; Lee, Y.S.; Singhal, N.S.; Scherer, P.E.; Lazar, M.A.; Ahima, R.S. Loss of resistin improves glucose homeostasis in leptin deficiency. Diabetes 2006, 55, 3083–3090. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Nguyen, M.T.; Miles, P.D.; Imamura, T.; Usui, I.; Olefsky, J.M. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J. Clin. Investig. 2004, 114, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by resistin. Mol. Cell. Biol. 2005, 25, 1569–1575. [Google Scholar] [CrossRef]
- Qatanani, M.; Szwergold, N.R.; Greaves, D.R.; Ahima, R.S.; Lazar, M.A. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J. Clin. Investig. 2009, 119, 531–539. [Google Scholar] [CrossRef]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem. Biophys. Res. Commun. 2003, 300, 472–476. [Google Scholar] [CrossRef]
- Mertens, I.; Van Gaal, L.F. Obesity, haemostasis and the fibrinolytic system. Obes. Rev. 2002, 3, 85–101. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Alessi, M.C.; Mavri, A.; Morange, P.E. Plasminogen activator inhibitor-1, inflammation, obesity, insulin resistance and vascular risk. J. Thromb. Haemost. 2003, 1, 1575–1579. [Google Scholar] [CrossRef]
- Ma, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.; Zhang, Y.; Brown, N.J.; Swift, L.L.; McGuinness, O.P.; Wasserman, D.H.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004, 53, 336–346. [Google Scholar] [CrossRef]
- Liang, X.; Kanjanabuch, T.; Mao, S.L.; Hao, C.M.; Tang, Y.W.; Declerck, P.J.; Hasty, A.H.; Wasserman, D.H.; Fogo, A.B.; Ma, L.J. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E103–E113. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, H.; Wang, Z.; Xiao, W. Plasminogen activator inhibitor-1 promotes inflammatory process induced by cigarette smoke extraction or lipopolysaccharides in alveolar epithelial cells. Exp. Lung Res. 2009, 35, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Bodles, A.M.; Phanavanh, B.; Lee, M.J.; Starks, T.; Kern, L.M.; Spencer, H.J., 3rd; McGehee, R.E., Jr.; et al. Human visfatin expression: Relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J. Clin. Endocrinol. Metab. 2007, 92, 666–672. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Saddi-Rosa, P.; Oliveira, C.S.; Giuffrida, F.M.; Reis, A.F. Visfatin, glucose metabolism and vascular disease: A review of evidence. Diabetol. Metab. Syndr. 2010, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chang, T.J.; Lee, W.J.; Chuang, L.M. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism 2010, 59, 93–99. [Google Scholar] [CrossRef]
- Haider, D.G.; Schindler, K.; Schaller, G.; Prager, G.; Wolzt, M.; Ludvik, B. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J. Clin. Endocrinol. Metab. 2006, 91, 1578–1581. [Google Scholar] [CrossRef]
- Oki, K.; Yamane, K.; Kamei, N.; Nojima, H.; Kohno, N. Circulating visfatin level is correlated with inflammation, but not with insulin resistance. Clin. Endocrinol. 2007, 67, 796–800. [Google Scholar] [CrossRef]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Kloting, N.; Graham, T.E.; Berndt, J.; Kralisch, S.; Kovacs, P.; Wason, C.J.; Fasshauer, M.; Schon, M.R.; Stumvoll, M.; Bluher, M.; et al. Serum retinol-binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metab. 2007, 6, 79–87. [Google Scholar] [CrossRef]
- Ost, A.; Danielsson, A.; Liden, M.; Eriksson, U.; Nystrom, F.H.; Stralfors, P. Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEB J. 2007, 21, 3696–3704. [Google Scholar] [CrossRef]
- Balagopal, P.; Graham, T.E.; Kahn, B.B.; Altomare, A.; Funanage, V.; George, D. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: Association with subclinical inflammation. J. Clin. Endocrinol. Metab. 2007, 92, 1971–1974. [Google Scholar] [CrossRef]
- Tabata, M.; Kadomatsu, T.; Fukuhara, S.; Miyata, K.; Ito, Y.; Endo, M.; Urano, T.; Zhu, H.J.; Tsukano, H.; Tazume, H.; et al. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab. 2009, 10, 178–188. [Google Scholar] [CrossRef]
- Ouchi, N.; Higuchi, A.; Ohashi, K.; Oshima, Y.; Gokce, N.; Shibata, R.; Akasaki, Y.; Shimono, A.; Walsh, K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 2010, 329, 454–457. [Google Scholar] [CrossRef]
- Tan, X.; Wang, X.; Chu, H.; Liu, H.; Yi, X.; Xiao, Y. SFRP5 correlates with obesity and metabolic syndrome and increases after weight loss in children. Clin. Endocrinol. 2014, 81, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, K.; Fuller, G.M.; Fuentes, N.L.; Moser, A.H.; Staprans, I.; Grunfeld, C.; Feingold, K.R. Interleukin-6 stimulates hepatic triglyceride secretion in rats. Endocrinology 1995, 136, 2143–2149. [Google Scholar] [CrossRef]
- Poitou, C.; Viguerie, N.; Cancello, R.; De Matteis, R.; Cinti, S.; Stich, V.; Coussieu, C.; Gauthier, E.; Courtine, M.; Zucker, J.D.; et al. Serum amyloid A: Production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 2005, 48, 519–528. [Google Scholar] [CrossRef]
- Sack, G.H., Jr. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Wang, P.H.; Liu, B.H.; Hsu, H.H.; Mersmann, H.J.; Ding, S.T. Serum amyloid A protein regulates the expression of porcine genes related to lipid metabolism. J. Nutr. 2008, 138, 674–679. [Google Scholar] [CrossRef]
- Cai, L.; de Beer, M.C.; de Beer, F.C.; van der Westhuyzen, D.R. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 2005, 280, 2954–2961. [Google Scholar] [CrossRef]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 2003, 24, 357–387. [Google Scholar] [CrossRef]
- Lewis, K.E.; Kirk, E.A.; McDonald, T.O.; Wang, S.; Wight, T.N.; O’Brien, K.D.; Chait, A. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation 2004, 110, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Berk, P.D. Lipid metabolism in hepatic steatosis. Clin. Liver Dis. 2004, 8, 639–671. [Google Scholar] [CrossRef]
- Koteish, A.; Diehl, A.M. Animal models of steatosis. Semin. Liver Dis. 2001, 21, 89–104. [Google Scholar] [CrossRef]
- Heilbronn, L.; Smith, S.R.; Ravussin, E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int. J. Obes. Relat. Metab. Disord. 2004, 28 (Suppl. S4), S12–S21. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bermejo, A.; Botas, P.; Funahashi, T.; Delgado, E.; Kihara, S.; Ricart, W.; Fernandez-Real, J.M. Adiponectin, hepatocellular dysfunction and insulin sensitivity. Clin. Endocrinol. 2004, 60, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Scala, L.; Poli, F.; Zenari, L.; Falezza, G. Decreased plasma adiponectin concentrations are closely associated with nonalcoholic hepatic steatosis in obese individuals. Clin. Endocrinol. 2004, 61, 700–703. [Google Scholar] [CrossRef]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.F.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Marra, F. Leptin and liver fibrosis: A matter of fat. Gastroenterology 2002, 122, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Mak, K.M.; Ren, C.; Lieber, C.S. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: Respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J. Biol. Chem. 2004, 279, 4292–4304. [Google Scholar] [CrossRef]
- Saxena, N.K.; Titus, M.A.; Ding, X.; Floyd, J.; Srinivasan, S.; Sitaraman, S.V.; Anania, F.A. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: Mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004, 18, 1612–1614. [Google Scholar] [CrossRef]
- Tobe, K.; Ogura, T.; Tsukamoto, C.; Imai, A.; Matsuura, K.; Iwasaki, Y.; Shimomura, H.; Higashi, T.; Tsuji, T. Relationship between serum leptin and fatty liver in Japanese male adolescent university students. Am. J. Gastroenterol. 1999, 94, 3328–3335. [Google Scholar] [CrossRef]
- Banerjee, R.R.; Lazar, M.A. Resistin: Molecular history and prognosis. J. Mol. Med. 2003, 81, 218–226. [Google Scholar] [CrossRef]
- Pagano, C.; Soardo, G.; Pilon, C.; Milocco, C.; Basan, L.; Milan, G.; Donnini, D.; Faggian, D.; Mussap, M.; Plebani, M.; et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J. Clin. Endocrinol. Metab. 2006, 91, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J. Cell. Mol. Med. 2004, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Marcellini, M.; Giannone, G.; Nobili, V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am. J. Clin. Pathol. 2007, 127, 954–960. [Google Scholar] [CrossRef]
- Saleh, J.; Christou, N.; Cianflone, K. Regional specificity of ASP binding in human adipose tissue. Am. J. Physiol. 1999, 276, E815–E821. [Google Scholar] [CrossRef] [PubMed]
- Massiera, F.; Bloch-Faure, M.; Ceiler, D.; Murakami, K.; Fukamizu, A.; Gasc, J.M.; Quignard-Boulange, A.; Negrel, R.; Ailhaud, G.; Seydoux, J.; et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001, 15, 2727–2729. [Google Scholar] [CrossRef]
- Umemura, S.; Nyui, N.; Tamura, K.; Hibi, K.; Yamaguchi, S.; Nakamaru, M.; Ishigami, T.; Yabana, M.; Kihara, M.; Inoue, S.; et al. Plasma angiotensinogen concentrations in obese patients. Am. J. Hypertens. 1997, 10, 629–633. [Google Scholar] [CrossRef]
- Borem, L.M.A.; Neto, J.F.R.; Brandi, I.V.; Lelis, D.F.; Santos, S.H.S. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: A brief review. Hypertens. Res. 2018, 41, 394–405. [Google Scholar] [CrossRef]
- Nair, K.S.; Short, K.R. Hormonal and signaling role of branched-chain amino acids. J. Nutr. 2005, 135 (Suppl. S6), 1547S–1552S. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef]
- Bloomgarden, Z. Diabetes and branched-chain amino acids: What is the link? J. Diabetes 2018, 10, 350–352. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1552–E1563. [Google Scholar] [CrossRef]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E311–E319. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef]
- Adams, T.D.; Gress, R.E.; Smith, S.C.; Halverson, R.C.; Simper, S.C.; Rosamond, W.D.; Lamonte, M.J.; Stroup, A.M.; Hunt, S.C. Long-term mortality after gastric bypass surgery. N. Engl. J. Med. 2007, 357, 753–761. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef]
- Jonasson, J.M.; Ljung, R.; Talback, M.; Haglund, B.; Gudbjornsdottir, S.; Steineck, G. Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia 2009, 52, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Girnun, G.D.; Chen, L.; Silvaggi, J.; Drapkin, R.; Chirieac, L.R.; Padera, R.F.; Upadhyay, R.; Vafai, S.B.; Weissleder, R.; Mahmood, U.; et al. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin. Cancer Res. 2008, 14, 6478–6486. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, R.; Ratnasinghe, L.; Simmons, D.L.; Siegel, E.R.; Midathada, M.V.; Kim, L.; Kim, P.J.; Owens, R.J.; Lang, N.P. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J. Clin. Oncol. 2007, 25, 1476–1481. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Haspinger, E.; La Russa, F.; Maspero, F.; Graziano, P.; Kovalszky, I.; Lovas, S.; Nama, K.; Hoffmann, R.; Knappe, D.; et al. Design and development of a peptide-based adiponectin receptor agonist for cancer treatment. BMC Biotechnol. 2011, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Smith, T.; Rahman, K.; Thorn, N.E.; Anania, F.A. Adiponectin agonist ADP355 attenuates CCl4-induced liver fibrosis in mice. PLoS ONE 2014, 9, e110405. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef]
- Philp, L.K.; Rockstroh, A.; Lehman, M.; Sadowski, M.C.; Bartonicek, N.; Wade, J.D.; Otvos, L.; Nelson, C.C. Adiponectin receptor activation inhibits prostate cancer xenograft growth. Endocr. Relat. Cancer 2020, 27, 711–729. [Google Scholar] [CrossRef]
- Sun, Y.; Zang, Z.; Zhong, L.; Wu, M.; Su, Q.; Gao, X.; Zan, W.; Lin, D.; Zhao, Y.; Zhang, Z. Identification of adiponectin receptor agonist utilizing a fluorescence polarization based high throughput assay. PLoS ONE 2013, 8, e63354. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Murakami, H.; Ono, A.; Takahashi, T.; Onozawa, Y.; Tsushima, T.; Yamazaki, K.; Jikoh, T.; Boku, N.; Yamamoto, N. Phase I study of Efatutazone, an oral PPARgamma agonist, in patients with metastatic solid tumors. Anticancer Res. 2014, 34, 5133–5141. [Google Scholar]
- Ory, V.; Kietzman, W.B.; Boeckelman, J.; Kallakury, B.V.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The PPARgamma agonist efatutazone delays invasive progression and induces differentiation of ductal carcinoma in situ. Breast Cancer Res. Treat. 2018, 169, 47–57. [Google Scholar] [CrossRef]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K.; Nagaretani, H.; Matsuda, M.; Komuro, R.; Ouchi, N.; et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef]
- Catalano, S.; Mauro, L.; Bonofiglio, D.; Pellegrino, M.; Qi, H.; Rizza, P.; Vizza, D.; Bossi, G.; Ando, S. In vivo and in vitro evidence that PPARgamma ligands are antagonists of leptin signaling in breast cancer. Am. J. Pathol. 2011, 179, 1030–1040. [Google Scholar] [CrossRef]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. 2019, 23, 101–111. [Google Scholar] [CrossRef]
- Tajima, K.; Nakamura, A.; Shirakawa, J.; Togashi, Y.; Orime, K.; Sato, K.; Inoue, H.; Kaji, M.; Sakamoto, E.; Ito, Y.; et al. Metformin prevents liver tumorigenesis induced by high-fat diet in C57Bl/6 mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E987–E998. [Google Scholar] [CrossRef]
- Schulte, L.; Scheiner, B.; Voigtlander, T.; Koch, S.; Schweitzer, N.; Marhenke, S.; Ivanyi, P.; Manns, M.P.; Rodt, T.; Hinrichs, J.B.; et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019, 39, 714–726. [Google Scholar] [CrossRef]
- Jiang, N.; Sun, R.; Sun, Q. Leptin signaling molecular actions and drug target in hepatocellular carcinoma. Drug Des. Dev. Ther. 2014, 8, 2295–2302. [Google Scholar]
- Stefanou, N.; Papanikolaou, V.; Furukawa, Y.; Nakamura, Y.; Tsezou, A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 2010, 10, 442. [Google Scholar] [CrossRef]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef]
- Harmon, T.; Harbuzariu, A.; Lanier, V.; Lipsey, C.C.; Kirlin, W.; Yang, L.; Gonzalez-Perez, R.R. Nanoparticle-linked antagonist for leptin signaling inhibition in breast cancer. World J. Clin. Oncol. 2017, 8, 54–66. [Google Scholar] [CrossRef]
- Catalano, S.; Leggio, A.; Barone, I.; De Marco, R.; Gelsomino, L.; Campana, A.; Malivindi, R.; Panza, S.; Giordano, C.; Liguori, A.; et al. A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell. Mol. Med. 2015, 19, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Wang, D.; Zhang, H.; Yi, X.; Sun, X.; Shi, B.; Wu, H.; Wu, G.; Wang, X.; Shang, Y. Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res. 2004, 64, 5870–5875. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Vertino, P.M.; Anania, F.A.; Sharma, D. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem. 2007, 282, 13316–13325. [Google Scholar] [CrossRef]
- Soma, D.; Kitayama, J.; Yamashita, H.; Miyato, H.; Ishikawa, M.; Nagawa, H. Leptin augments proliferation of breast cancer cells via transactivation of HER2. J. Surg. Res. 2008, 149, 9–14. [Google Scholar] [CrossRef]
- Ho, Y.; Wang, S.H.; Chen, Y.R.; Li, Z.L.; Chin, Y.T.; Yang, Y.S.H.; Wu, Y.H.; Su, K.W.; Chu, H.R.; Chiu, H.C.; et al. Leptin-derived peptides block leptin-induced proliferation by reducing expression of pro-inflammatory genes in hepatocellular carcinoma cells. Food Chem. Toxicol. 2019, 133, 110808. [Google Scholar] [CrossRef]
- Erstad, D.J.; Tager, A.M.; Hoshida, Y.; Fuchs, B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell. Oncol. 2017, 4, e1311827. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective Inhibition of Autotaxin Is Efficacious in Mouse Models of Liver Fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Wei, L.; Song, W.M.; Higashi, T.; Ghoshal, S.; Kim, R.S.; Bian, C.B.; Yamada, S.; Sun, X.; Venkatesh, A.; et al. Molecular Liver Cancer Prevention in Cirrhosis by Organ. Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 2016, 30, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef]
- Trikha, M.; Corringham, R.; Klein, B.; Rossi, J.F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: A review of the rationale and clinical evidence. Clin. Cancer Res. 2003, 9, 4653–4665. [Google Scholar]
- Roxburgh, C.S.; McMillan, D.C. Therapeutics targeting innate immune/inflammatory responses through the interleukin-6/JAK/STAT signal transduction pathway in patients with cancer. Transl. Res. 2016, 167, 61–66. [Google Scholar] [CrossRef]
- Chen, R.; Chen, B. Siltuximab (CNTO 328): A promising option for human malignancies. Drug Des. Dev. Ther. 2015, 9, 3455–3458. [Google Scholar] [CrossRef]
- Ohsugi, Y. Recent advances in immunopathophysiology of interleukin-6: An innovative therapeutic drug, tocilizumab (recombinant humanized anti-human interleukin-6 receptor antibody), unveils the mysterious etiology of immune-mediated inflammatory diseases. Biol. Pharm. Bull. 2007, 30, 2001–2006. [Google Scholar] [CrossRef]
- Dechandt, C.R.P.; Zuccolotto-Dos-Reis, F.H.; Teodoro, B.G.; Fernandes, A.; Eberlin, M.N.; Kettelhut, I.C.; Curti, C.; Alberici, L.C. Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J. Bioenerg. Biomembr. 2017, 49, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.L.; He, Y.; Ji, L.L.; Wang, K.Y.; Wang, Y.L.; Chen, D.F.; Geng, Y.; OuYang, P.; Lai, W.M. Hepatoprotective potential of isoquercitrin against type 2 diabetes-induced hepatic injury in rats. Oncotarget 2017, 8, 101545–101559. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Robertson, C.L.; Ebeid, K.; Dozmorov, M.; Rajasekaran, D.; Mendoza, R.; Siddiq, A.; Akiel, M.A.; Jariwala, N.; Shen, X.N.; et al. A novel role of astrocyte elevated gene-1 (AEG-1) in regulating nonalcoholic steatohepatitis (NASH). Hepatology 2017, 66, 466–480. [Google Scholar] [CrossRef]
- Srivastava, J.; Siddiq, A.; Emdad, L.; Santhekadur, P.K.; Chen, D.; Gredler, R.; Shen, X.N.; Robertson, C.L.; Dumur, C.I.; Hylemon, P.B.; et al. Astrocyte elevated gene-1 promotes hepatocarcinogenesis: Novel insights from a mouse model. Hepatology 2012, 56, 1782–1791. [Google Scholar] [CrossRef]
- Robertson, C.L.; Srivastava, J.; Siddiq, A.; Gredler, R.; Emdad, L.; Rajasekaran, D.; Akiel, M.; Shen, X.N.; Guo, C.; Giashuddin, S.; et al. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res. 2014, 74, 6184–6193. [Google Scholar] [CrossRef]
- Rajasekaran, D.; Srivastava, J.; Ebeid, K.; Gredler, R.; Akiel, M.; Jariwala, N.; Robertson, C.L.; Shen, X.N.; Siddiq, A.; Fisher, P.B.; et al. Combination of Nanoparticle-Delivered siRNA for Astrocyte Elevated Gene-1 (AEG-1) and All-trans Retinoic Acid (ATRA): An Effective Therapeutic Strategy for Hepatocellular Carcinoma (HCC). Bioconjug. Chem. 2015, 26, 1651–1661. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).