
Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
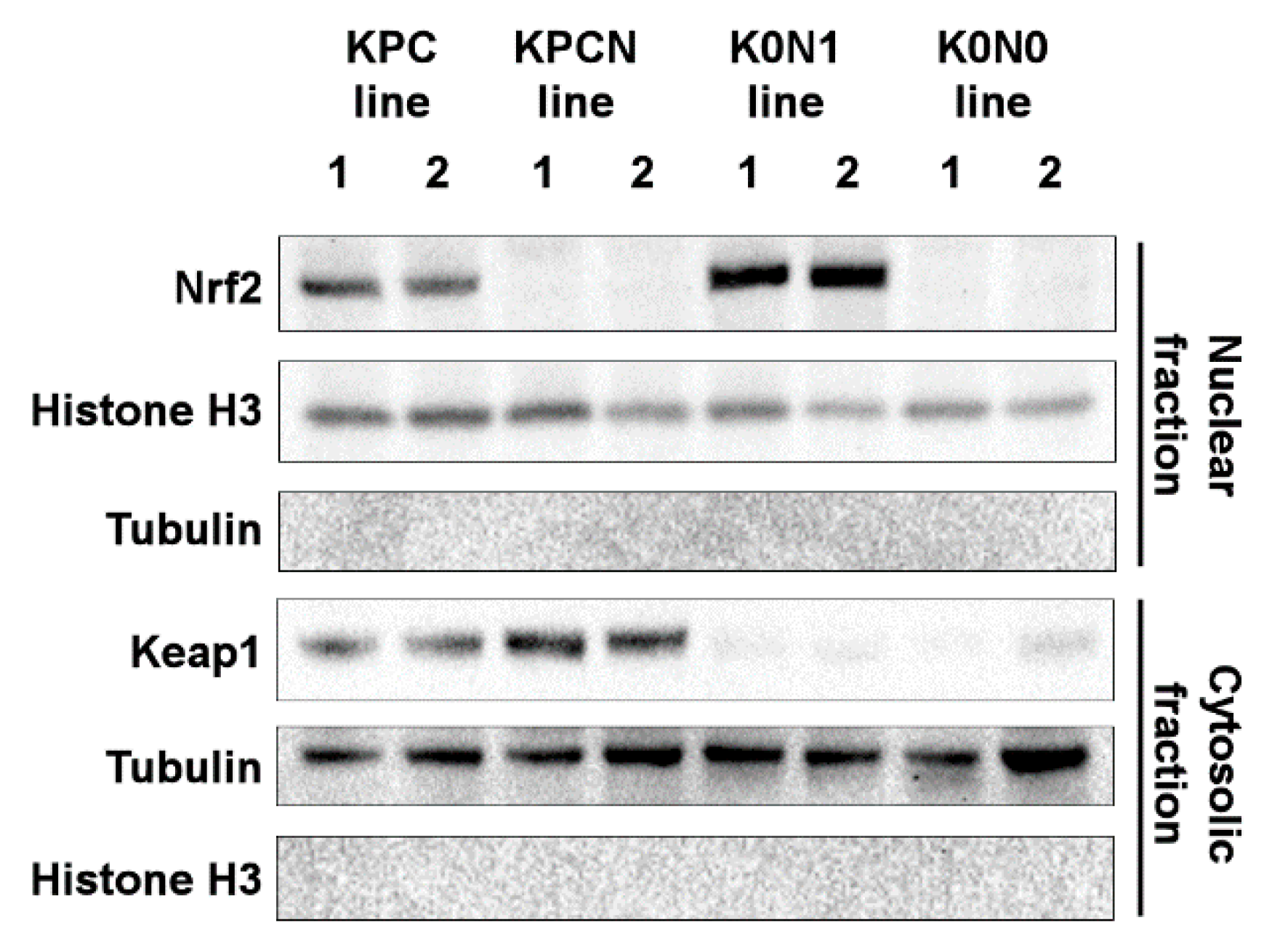
2.1. Establishment of Cell Lines Expressing Constitutively Activated Nrf2
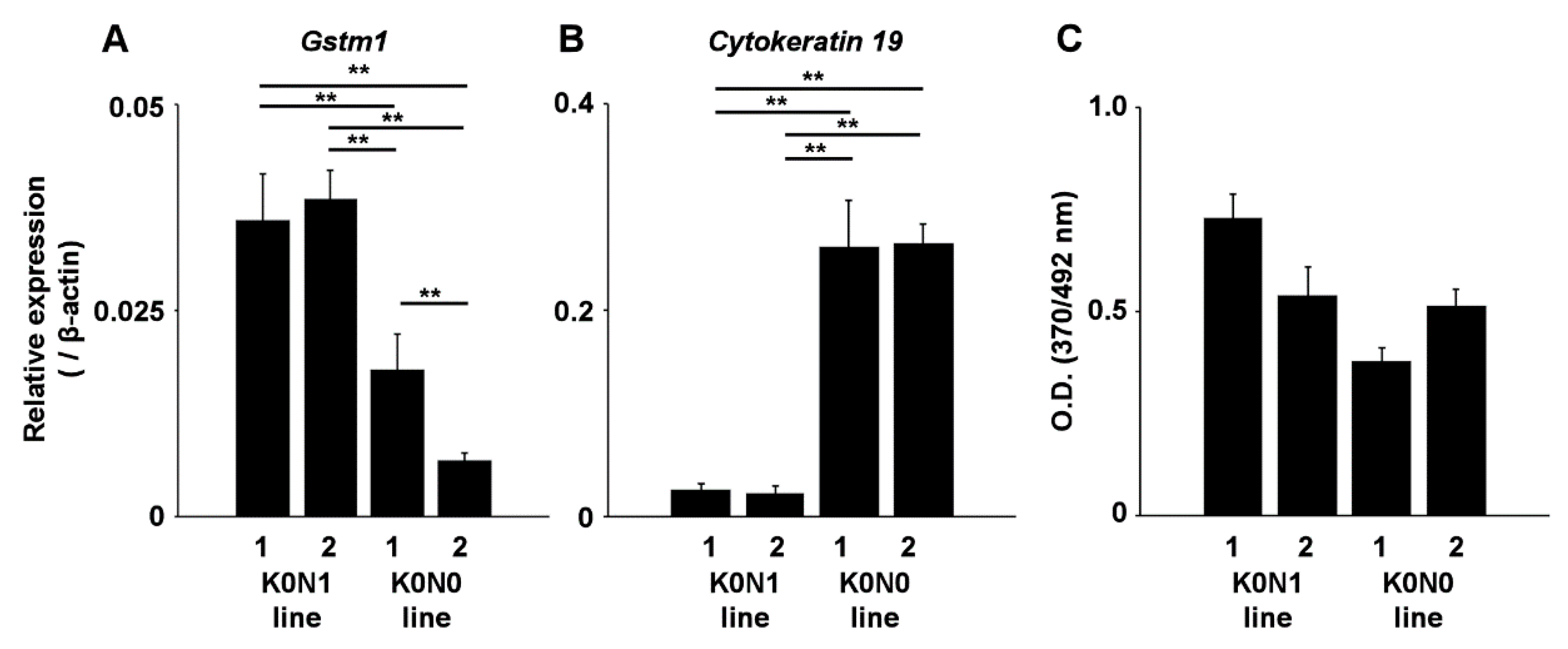
2.2. Increased Expression of Nrf2-Target Genes in Cell Lines Expressing Constitutively Activated Nrf2
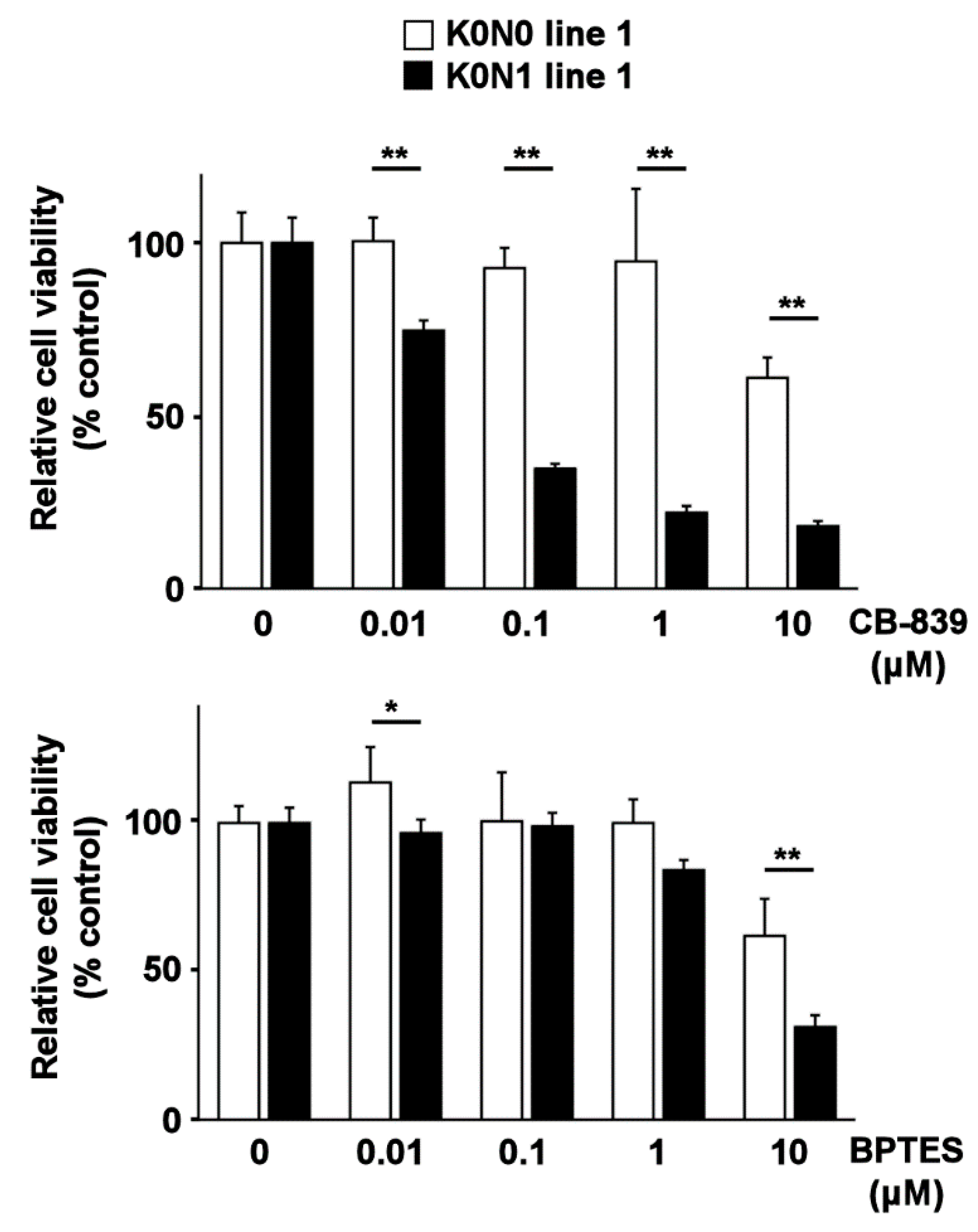
2.3. Cell Lines Expressing Constitutively Activated Nrf2 Are Sensitive to Glutaminase Inhibitors
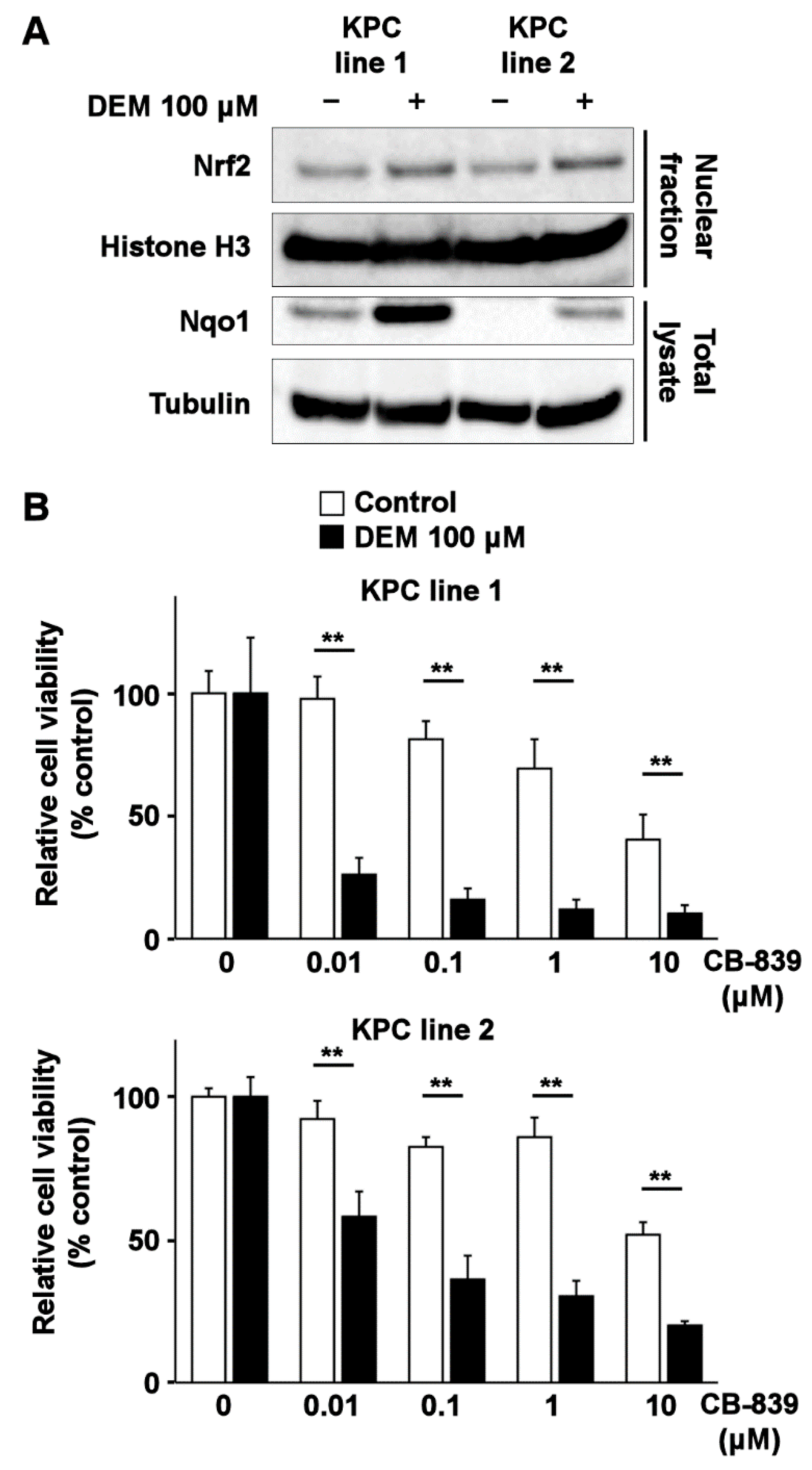
2.4. Nrf2 Inducer Sensitizes Murine Pancreatic Cancer Cell Lines to Glutaminase Inhibitor
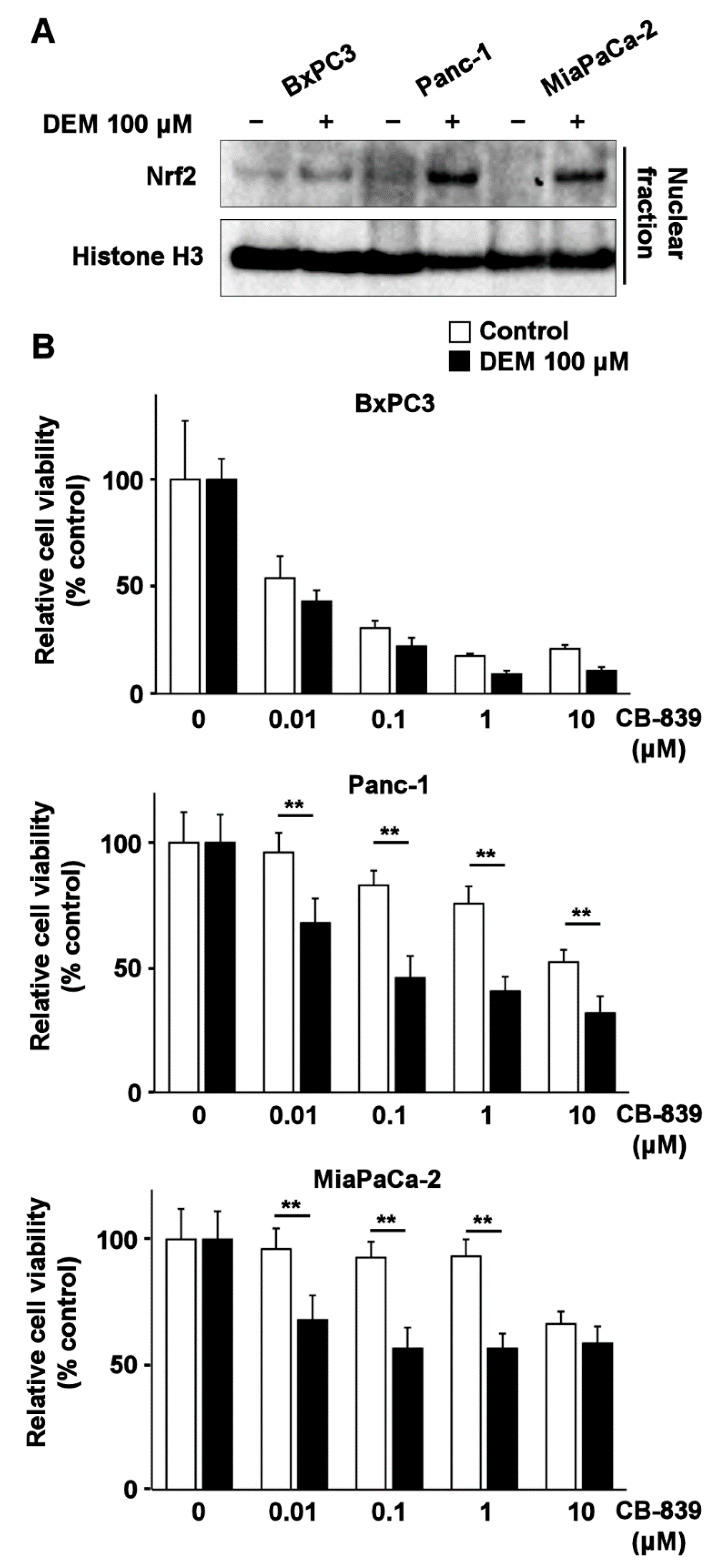
2.5. Nrf2 Inducer Sensitizes Human Pancreatic Cancer Cell Lines Harboring K-Ras Mutation to Glutaminase Inhibitor
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mice
4.3. Cell Lines and Cell Culture
4.4. RNA Extraction and Quantitative RT-PCR
4.5. Western Blot
4.6. Cell Growth Assay
4.7. Cell Viability Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dematteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 1097–1104. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Lin, H.; Zhang, X.; Fan, Q.; Chen, S.; Shahda, S.; Liu, Y.; Sun, J.; Xie, J. Simultaneous inhibition of MEK and Hh signaling reduces pancreatic cancer metastasis. Cancers 2018, 10, 403. [Google Scholar] [CrossRef]
- Shen, L.; Kim, S.H.; Chen, C.Y. Sensitization of human pancreatic cancer cells harboring mutated K-ras to apoptosis. PLoS ONE 2012, 7, e40435. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; Konig, K.; Michels, S.; Fischer, R.; et al. Clinical and pathological characteristics of KEAP1- and NFE2L2-mutated non-small cell lung carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Feng, X.; Chen, Y.; Selfridge, J.E.; Gorityala, S.; Du, Z.; Wang, J.M.; Hao, Y.; Cioffi, G.; Conlon, R.A.; et al. 5-Fluorouracil enhances the antitumor activity of the glutaminase inhibitor CB-839 against PIK3CA-mutant colorectal cancers. Cancer Res. 2020, 80, 4815–4827. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martin-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Hamada, S.; Shimosegawa, T.; Taguchi, K.; Nabeshima, T.; Yamamoto, M.; Masamune, A. Simultaneous K-ras activation and Keap1 deletion cause atrophy of pancreatic parenchyma. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G65–G74. [Google Scholar] [CrossRef]
- Nabeshima, T.; Hamada, S.; Taguchi, K.; Tanaka, Y.; Matsumoto, R.; Yamamoto, M.; Masamune, A. Keap1 deletion accelerates mutant K-ras/p53-driven cholangiocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G419–G427. [Google Scholar] [CrossRef]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Lozanovski, V.J.; Houben, P.; Hinz, U.; Hackert, T.; Herr, I.; Schemmer, P. Pilot study evaluating broccoli sprouts in advanced pancreatic cancer (POUDER trial)—Study protocol for a randomized controlled trial. Trials 2014, 15, 204. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, C.; Carter, J.L. An evaluation of dimethyl fumarate for the treatment of relapsing remitting multiple sclerosis. Expert Opin. Pharmacother. 2020, 21, 1399–1405. [Google Scholar] [CrossRef]
- Maruyama, A.; Tsukamoto, S.; Nishikawa, K.; Yoshida, A.; Harada, N.; Motojima, K.; Ishii, T.; Nakane, A.; Yamamoto, M.; Itoh, K. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch. Biochem. Biophys. 2008, 477, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; McEvoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 2007, 12, 1163–1178. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Wang, X.; Spandidos, A.; Wang, H.; Seed, B. PrimerBank: A PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012, 40, D1144–D1149. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamada, S.; Matsumoto, R.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. Int. J. Mol. Sci. 2021, 22, 1870. https://doi.org/10.3390/ijms22041870
Hamada S, Matsumoto R, Tanaka Y, Taguchi K, Yamamoto M, Masamune A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. International Journal of Molecular Sciences. 2021; 22(4):1870. https://doi.org/10.3390/ijms22041870
Chicago/Turabian StyleHamada, Shin, Ryotaro Matsumoto, Yu Tanaka, Keiko Taguchi, Masayuki Yamamoto, and Atsushi Masamune. 2021. "Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition" International Journal of Molecular Sciences 22, no. 4: 1870. https://doi.org/10.3390/ijms22041870
APA StyleHamada, S., Matsumoto, R., Tanaka, Y., Taguchi, K., Yamamoto, M., & Masamune, A. (2021). Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. International Journal of Molecular Sciences, 22(4), 1870. https://doi.org/10.3390/ijms22041870