
Chemical Constituents of the Bulbs of Scilla peruviana and Their Pancreatic Lipase Inhibitory Activity

Abstract
1. Introduction
2. Results and Discussion
2.1. Structural Elucidation
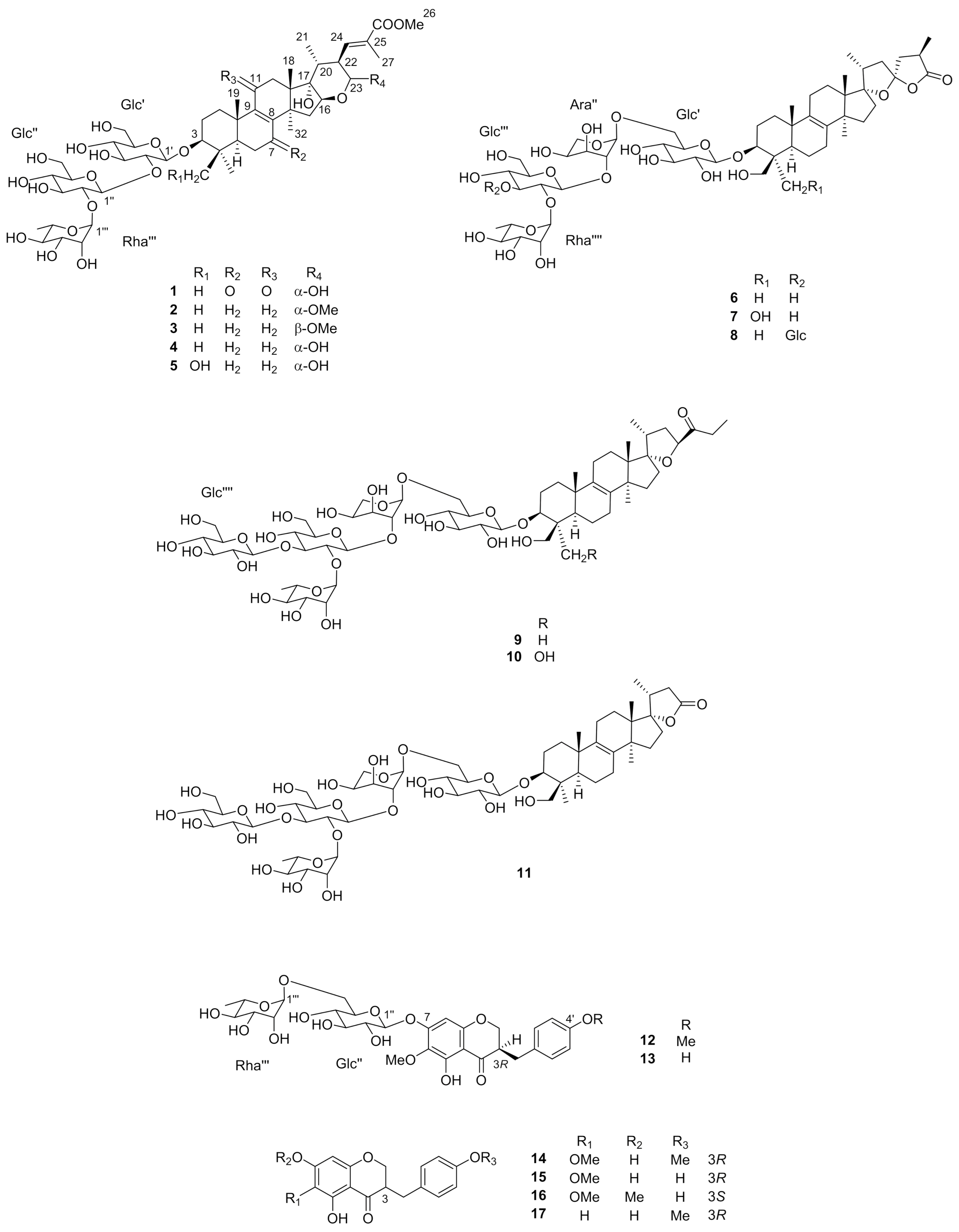
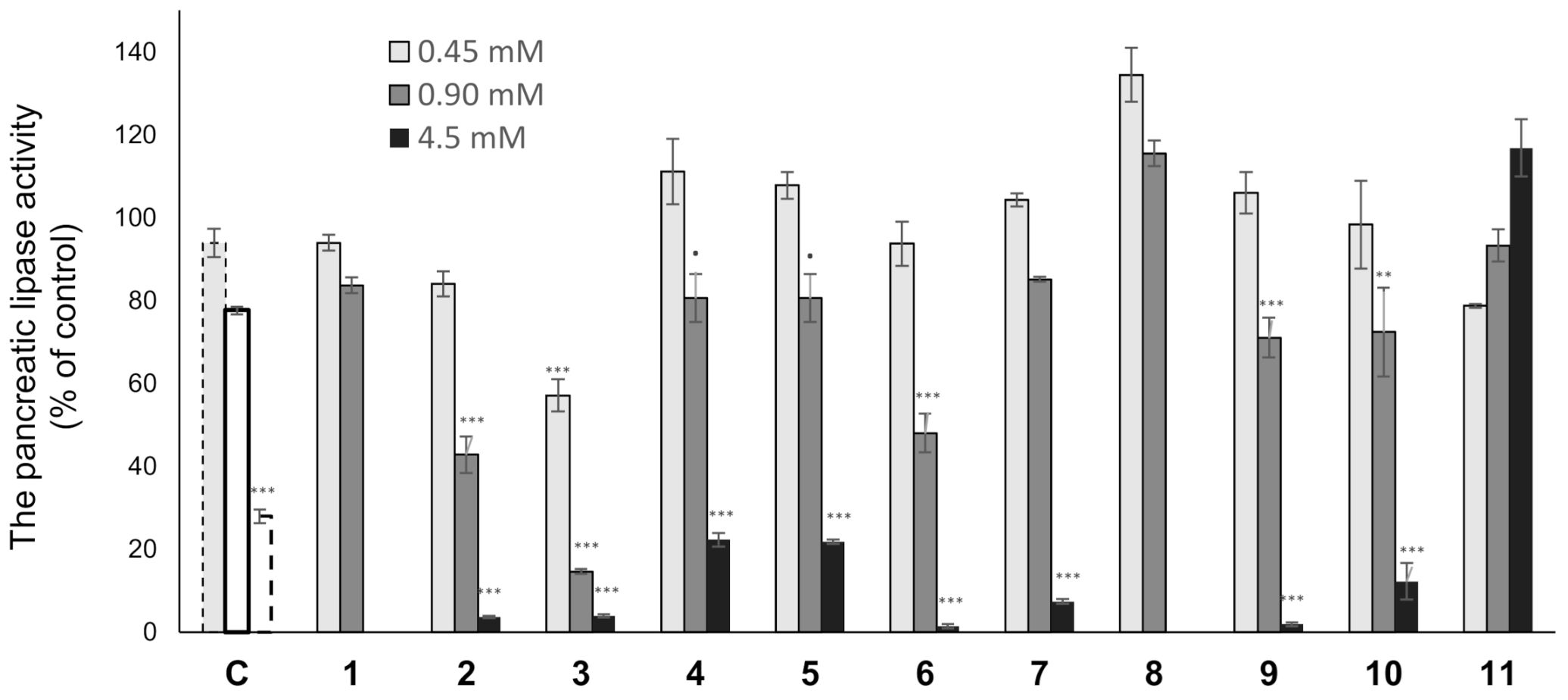
2.2. Lipase Inhibitory Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Structural Characterization
3.5. Lipase-Inhibitory Activity
3.6. Animals
3.7. Measurement of Triglyceride in Animals
3.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ori, K.; Kuroda, M.; Mimaki, Y.; Sakagami, H.; Sashida, Y. Norlanostane and lanostane glycosides from the bulbs of Chionodoxa luciliae and their cytotoxic activity. Chem. Pharm. Bull. 2003, 51, 92–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuroda, M.; Mimaki, Y.; Ori, K.; Koshino, H.; Nukada, T.; Sakagami, H.; Sashida, Y. Lucilianosides A and B, two novel tetranor-lanostane hexaglycosides from the bulbs of Chionodoxa luciliae. Tetrahedron 2002, 58, 6735–6740. [Google Scholar] [CrossRef]
- Ori, K.; Kuroda, M.; Mimaki, Y.; Sakagami, H.; Sashida, Y. Lanosterol and tetranorlanosterol glycosides from the bulbs of Muscari paradoxum. Phytochemistry 2003, 64, 1351–1359. [Google Scholar] [CrossRef]
- Lee, H.B.; Lee, S.M. Antimicrobial activity of eucosterol oligosaccharides isolated from bulb of squill (Scilla scilloides). Pharmacol. Pharm. 2013, 4, 110–114. [Google Scholar] [CrossRef]
- Ono, M.; Toyohisa, D.; Morishita, T.; Horita, H.; Yasuda, S.; Nishida, Y.; Nohara, T. Three new nortriterpene glycosides and two new triterpene glycosides from the bulbs of Scilla scilloides. Chem. Pharm. Bull. 2011, 59, 1348–1354. [Google Scholar] [CrossRef][Green Version]
- Nishida, Y.; Sugahara, S.; Wada, K.; Toyohisa, D.; Tanaka, T.; Ono, M.; Yasuda, S. Inhibitory effects of the ethyl acetate extract from bulbs of Scilla scilloides on lipoxygenase and hyaluronidase activities. Pharm. Biol. 2014, 52, 1351–1357. [Google Scholar] [CrossRef]
- Mimaki, Y.; Ori, K.; Sashida, Y.; Nikaido, T.; Song, L.G.; Ohmoto, T. Peruvianosides A and B, novel triterpene glycosides from the bulbs of Scilla peruviana. Bull. Chem. Soc. Jpn. 1993, 66, 1182–1186. [Google Scholar] [CrossRef]
- Mimaki, Y.; Ori, K.; Sashida, Y.; Nikaido, T.; Song, L.G. Peruvianoside A, a novel migrated lanostane trisaccharide from Scilla peruviana. Chem. Lett. 1992, 10, 1999–2000. [Google Scholar] [CrossRef]
- Sholichin, M.; Miyahara, K.; Kawasaki, T. Oligoglycosides of spirocyclic nortriterpenoids related to eucosterol. Chem. Pharm. Bull. 1985, 33, 1756–1759. [Google Scholar] [CrossRef][Green Version]
- Mimaki, Y.; Nishino, H.; Ori, K.; Kuroda, M.; Matsui, T.; Sashida, Y. Lanosterol oligosaccharides from the plants of the subfamily Scilloideae and their antitumor-promoter activity. Chem. Pharm. Bull. 1994, 42, 327–332. [Google Scholar] [CrossRef]
- Mimaki, Y.; Ori, K.; Kubo, S.; Sashida, Y.; Nikaido, T.; Song, L.G.; Ohmoto, T. Scillasaponins A, B, and C, new triterpenoid oligosaccharides from the plants of the subfamily Scilloideae. Chem. Lett. 1992, 21, 1863–1866. [Google Scholar] [CrossRef]
- Morikawa, T.; Xuezheng, L.; Nishida, E.; Nakamura, S.; Ninomiya, K.; Matsuda, H.; Yoshikawa, M. Acylated oleanane-type triterpene bisdesmosides: Perennisaponins G, H, I, J, K, L, and M with pancreatic lipase inhibitory activity from the flowers of Bellis perennis. Helv. Chim. Acta 2010, 93, 573–586. [Google Scholar] [CrossRef]
- Jang, D.S.; Lee, G.Y.; Kim, J.; Lee, Y.M.; Kim, J.M.; Kim, Y.S.; Kim, J.S. A new pancreatic lipase inhibitor isolated from the roots of Actinidia arguta. Arch. Pharmacal Res. 2008, 31, 666–670. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, W.; Han, L.; Koike, K. Pancreatic lipase-inhibiting triterpenoid saponins from Gypsophila oldhamiana. Chem. Pharm. Bull. 2007, 55, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kubota, A.; Masimo, R.; Fukaya, H.; Inaba, N.; Mimaki, Y. Cymbopogonol from Cymbopogon citratus and its biological activity. Shoyakugaku Zasshi 2017, 71, 98–99. [Google Scholar]
- de la Garza, A.L.; Milagro-Yoldi, F.I.; Boque, N.; Campión-Zabalza, J.; Martinez, J.A. Natural inhibitors of pancreatic lipase as new players in obesity treatment. Planta Med. 2011, 77, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Chun, H.K.; Lee, C.H.; Min, B.S.; Lee, E.S.; Kho, Y.H. Eucosterol oligoglycosides isolated from Scilla scilloides and their anti-tumor activity. Chem. Pharm. Bull. 2002, 50, 1245–1249. [Google Scholar] [CrossRef]
- Ono, M.; Takatsu, Y.; Ochiai, T.; Yasuda, S.; Nishida, Y.; Tanaka, T.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T. Two new nortriterpenoid glycosides and a new phenylpropanoid glycoside from the bulbs of Scilla scilloides. Chem. Pharm. Bull. 2012, 60, 1314–1319. [Google Scholar] [CrossRef][Green Version]
- Crouch, N.R.; Bangani, V.; Mulholland, D.A. Homoisoflavanones from three South African: Scilla species. Phytochemistry 1999, 51, 943–946. [Google Scholar] [CrossRef]
- Adinolfi, M.; Lanzetta, R.; Laonigro, G.; Parrilli, M.; Breitmaier, E. 1H and 13C chemical shift assignments of homoisoflavanones. Magn. Reson. Chem. 1986, 24, 663–666. [Google Scholar] [CrossRef]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Morales-Serna, J.A.; Jiménez, A.; Estrada-Reyes, R.; Marquez, C.; Cárdenas, J.; Salmón, M. Homoisoflavanones from Agave tequilana weber. Molecules 2010, 15, 3295–3301. [Google Scholar] [CrossRef] [PubMed]
- Lunagariya, N.A.; Patel, N.K.; Jagtap, S.C.; Bhutani, K.K. Inhibitors of pancreatic lipase: State of the art and clinical perspectives. EXCLI J. 2014, 13, 897–921. [Google Scholar] [PubMed]
- Karu, N.; Reifen, R.; Kerem, Z. Weight gain reduction in mice fed Panax ginseng saponin, a pancreatic lipase inhibitor. J. Agric. Food Chem. 2007, 55, 2824–2828. [Google Scholar] [CrossRef]
- Yamada, Y.; Kato, T.; Ogino, H.; Ashina, S.; Kato, K. Cetilistat (ATL-962), a novel pancreatic lipase inhibitor, ameliorates body weight gain and improves lipid profiles in rats. Horm. Metab. Res. 2008, 40, 539–543. [Google Scholar] [CrossRef]
- Mosa, R.A.; Naidoo, J.J.; Nkomo, F.S.; Mazibuko, S.E.; Muller, C.J.F.; Opoku, A.R. In vitro anti-hyperlipidemic potential of triterpenes from stem bark of Protorhus longifolia. Planta Med. 2014, 80, 1685–1691. [Google Scholar]
- Jeepipalli, S.P.; Du, B.; Sabitaliyevich, U.Y.; Xu, B. New insights into potential nutritional effects of dietary saponins in protecting against the development of obesity. Food Chem. 2020, 318, 126474. [Google Scholar] [CrossRef]
- Matsuo, Y.; Takaku, R.; Mimaki, Y. Novel steroidal glycosides from the bulbs of Lilium pumilum. Molecules 2015, 20, 16255–16265. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | δH | J (Hz) | δC | δH | J (Hz) | δC | δH | J (Hz) | δC | |||
| 1a | 3.00 | dt | 13.8, 3.3 | 34.4 | 1.54 | m | 35.8 | 1.62 | m | 35.8 | ||
| b | 1.21 | br dd | 13.8, 3.4 | 1.10 | m | 1.19 | m | |||||
| 2a | 2.28 | m | 26.9 | 2.26 | m | 27.1 | 2.26 | m | 27.1 | |||
| b | 1.92 | m | 1.85 | m | 1.84 | m | ||||||
| 3 | 3.35 | dd | 11.8, 4.7 | 88.4 | 3.33 | dd | 11.3, 4.2 | 89.7 | 3.36 | dd | 11.7, 4.2 | 89.7 |
| 4 | — | 36.9 | — | 39.8 | — | 39.8 | ||||||
| 5 | 1.69 | br d | 7.8 | 50.7 | 1.15 | br d | 7.3 | 51.0 | 1.11 | br d | 7.8 | 51.1 |
| 6a | 2.60 | br d | 8.7 | 36.5 | 1.70 | dd | 12.1, 6.2 | 18.4 | 1.71 | dd | 12.0, 6.1 | 18.4 |
| b | 2.30 | m | 1.51 | m | 1.50 | m | ||||||
| 7 | — | 201.9 | 2.08 | m | 26.6 | 2.10 | m | 26.6 | ||||
| 1.51 | m | 1.39 | m | |||||||||
| 8 | — | 151.2 | — | 135.9 | — | 135.9 | ||||||
| 9 | — | 151.6 | — | 135.0 | — | 135.1 | ||||||
| 10 | — | 39.9 | — | 37.0 | — | 37.1 | ||||||
| 11 | — | 203.0 | 2.11 | m | 20.8 | 2.14 | m | 20.8 | ||||
| 2.00 | m | 2.06 | m | |||||||||
| 12a | 3.75 | d | 15.9 | 48.9 | 2.45 | dt | 13.0, 9.2 | 25.4 | 2.45 | m | 25.7 | |
| b | 2.77 | d | 15.9 | 1.58 | br dd | 13.0, 4.9 | 1.63 | br dd | 13.0, 3.4 | |||
| 13 | — | 52.4 | — | 48.5 | — | 48.6 | ||||||
| 14 | — | 47.4 | — | 48.1 | — | 48.3 | ||||||
| 15a | 3.36 | dd | 12.9, 7.4 | 42.5 | 2.22 | m | 44.4 | 2.23 | dd | 12.2, 7.8 | 44.4 | |
| b | 2.50 | dd | 12.9, 6.8 | 2.03 | m | 2.03 | m | |||||
| 16 | 5.10 | dd | 7.4, 6.8 | 79.4 | 5.06 | dd | 9.2, 6.2 | 80.2 | 4.52 | m | 81.6 | |
| 17 | — | 82.1 | — | 99.3 | — | 98.8 | ||||||
| 18 | 1.28 | s | 21.1 | 1.20 | s | 18.2 | 1.24 | s | 19.0 | |||
| 19 | 1.37 | s | 17.4 | 0.99 | s | 19.3 | 1.00 | s | 19.3 | |||
| 20 | 2.29 | dq | 11.8, 6.9 | 36.2 | 3.31 | dq | 10.0, 6.9 | 43.3 | 3.76 | dq | 8.6, 6.1 | 46.6 |
| 21 | 1.07 | d | 6.9 | 11.9 | 1.15 | d | 6.9 | 13.0 | 1.34 | d | 6.1 | 13.2 |
| 22 | 3.38 | ddd | 11.8, 10.6, 6.5 | 44.3 | 3.98 | ddd | 10.9, 10.0, 6.3 | 49.5 | 3.27 | ddd | 8.7, 8.6, 5.8 | 39.8 |
| 23 | 5.24 | d | 6.5 | 97.6 | 5.26 | d | 6.3 | 110.4 | 5.05 | d | 5.8 | 103.9 |
| 24 | 6.96 | dd | 10.6, 1.2 | 143.5 | 7.22 | dd | 10.9, 1.1 | 140.8 | 7.48 | dd | 8.7, 1.2 | 139.8 |
| 25 | — | 130.3 | — | 130.8 | — | 129.6 | ||||||
| 26 | — | 168.3 | — | 168.1 | — | 168.3 | ||||||
| 27 | 1.99 | d | 1.2 | 13.3 | 2.03 | d | 1.1 | 13.4 | 2.11 | d | 1.2 | 13.5 |
| 28 | — | — | — | — | — | — | ||||||
| 29 | — | — | — | — | — | — | ||||||
| 30 | 1.30 | s | 27.9 | 1.40 | s | 28.3 | 1.41 | s | 28.3 | |||
| 31 | 1.16 | s | 16.5 | 1.17 | s | 16.7 | 1.17 | s | 16.7 | |||
| 32 | 1.84 | s | 30.7 | 1.46 | s | 27.2 | 1.43 | s | 27.0 | |||
| 23-OMe | — | — | 3.49 | s | 56.4 | 3.42 | s | 55.1 | ||||
| 26-OMe | 3.71 | s | 51.6 | 3.71 | s | 51.7 | 3.67 | s | 51.6 |
| 1 | 2 | 3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | δH | J (Hz) | δC | δH | J (Hz) | δC | δH | J (Hz) | δC | ||||
| Glc | 1′ | 4.91 | d | 7.7 | 105.2 | 4.97 | d | 7.6 | 105.2 | 4.97 | d | 8.3 | 105.2 |
| 2′ | 4.42 | dd | 9.0, 7.7 | 78.7 | 4.43 | dd | 9.2, 7.6 | 79.0 | 4.44 | dd | 9.1, 8.3 | 79.0 | |
| 3′ | 4.54 | t | 9.0 | 79.5 | 4.56 | t | 9.2 | 79.5 | 4.56 | t | 9.1 | 79.5 | |
| 4′ | 4.06 | t | 9.0 | 71.9 | 4.10 | t | 9.2 | 72.0 | 4.10 | t | 9.1 | 71.9 | |
| 5′ | 3.89 | ddd | 9.0, 5.4, 2.7 | 77.9 | 3.94 | m | 77.9 | 3.93 | m | 77.9 | |||
| 6′a | 4.51 | dd | 11.9, 2.7 | 62.8 | 4.55 | dd | 11.7, 3.4 | 62.8 | 4.53 | br d | 11.4 | 62.8 | |
| b | 4.35 | dd | 11.9, 5.4 | 4.39 | dd | 11.7, 5.3 | 4.34 | dd | 11.4, 5.5 | ||||
| Glc | 1′′ | 5.83 | d | 6.9 | 101.9 | 5.85 | d | 7.5 | 102.0 | 5.86 | d | 7.4 | 102.0 |
| 2′′ | 4.24 | dd | 8.9, 6.9 | 78.7 | 4.31 | dd | 8.9, 7.5 | 78.6 | 4.33 | dd | 8.9, 7.4 | 78.6 | |
| 3′′ | 4.23 | t | 8.9 | 79.4 | 4.25 | t | 8.9 | 79.4 | 4.25 | t | 8.9 | 79.4 | |
| 4′′ | 4.06 | t | 8.9 | 72.9 | 4.09 | t | 8.9 | 72.9 | 4.09 | t | 8.9 | 72.9 | |
| 5′′ | 3.85 | ddd | 8.9, 5.7, 3.0 | 77.6 | 3.85 | ddd | 8.9, 5.3, 2.6 | 77.5 | 3.86 | ddd | 8.9, 5.8, 3.1 | 77.5 | |
| 6′′a | 4.49 | dd | 10.8, 3.0 | 63.5 | 4.49 | dd | 11.3, 2.6 | 63.5 | 4.49 | dd | 11.5, 3.1 | 63.5 | |
| b | 4.29 | dd | 10.8, 5.7 | 4.31 | dd | 11.3, 5.3 | 4.31 | dd | 11.5, 5.8 | ||||
| Rha | 1′′′ | 6.38 | br s | 102.1 | 6.41 | br s | 102.1 | 6.41 | br s | 102.1 | |||
| 2′′′ | 4.76 | br d | 3.3 | 72.4 | 4.76 | br d | 3.3 | 72.4 | 4.76 | br d | 3.3 | 72.4 | |
| 3′′′ | 4.67 | dd | 9.3, 3.3 | 72.7 | 4.69 | dd | 9.2, 3.3 | 72.7 | 4.69 | dd | 9.3, 3.3 | 72.7 | |
| 4′′′ | 4.32 | t | 9.3 | 74.3 | 4.32 | t | 9.2 | 74.3 | 4.33 | t | 9.3 | 74.3 | |
| 5′′′ | 5.01 | dq | 9.3, 6.3 | 69.5 | 5.06 | dq | 9.2, 6.2 | 69.6 | 5.07 | dq | 9.3, 6.2 | 69.6 | |
| 6′′′ | 1.74 | d | 6.3 | 19.0 | 1.80 | d | 6.2 | 18.9 | 1.80 | d | 6.2 | 19.0 | |
| 12 | 13 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C | δH | J (Hz) | δC | δH | J (Hz) | δC | |||
| 2 | 4.36 | dd | 11.5, 4.0 | 70.8 | 4.35 | dd | 11.5, 4.5 | 71.4 | |
| 4.19 | dd | 11.5, 6.5 | 4.20 | dd | 11.5, 7.0 | ||||
| 3 | 2.96 | m | 48.2 | 2.96 | m | 49.4 | |||
| 4 | — | 200.0 | — | 201.7 | |||||
| 4 | a | — | 101.8 | — | 105.2 | ||||
| 5 | — | 155.7 | — | 156.4 | |||||
| 6 | — | 131.2 | — | 132.0 | |||||
| 7 | — | 160.8 | — | 160.8 | |||||
| 8 | 6.31 | s | 97.4 | 6.31 | s | 98.1 | |||
| 8 | a | — | 160.1 | — | 162.2 | ||||
| 9 | 3.13 | dd | 14.0, 9.0 | 33.0 | 3.09 | dd | 14.0, 9.5 | 33.9 | |
| 2.72 | dd | 14.0, 7.0 | 2.68 | dd | 14.0, 7.3 | ||||
| 1′ | — | 131.2 | — | 132.0 | |||||
| 2′ | 7.16 | d | 8.5 | 131.2 | 7.06 | d | 8.5 | 132.0 | |
| 3′ | 6.86 | d | 8.5 | 115.2 | 6.73 | d | 8.5 | 117.2 | |
| 4′ | — | 160.0 | — | 158.3 | |||||
| 5′ | 6.86 | d | 8.5 | 115.1 | 6.73 | d | 8.5 | 117.2 | |
| 6′ | 7.16 | d | 8.5 | 131.2 | 7.06 | d | 8.5 | 132.0 | |
| 6-OCH3 | 3.75 | s | 61.4 | 3.75 | s | 62.7 | |||
| 4′-OCH3 | 3.77 | s | 55.7 | — | |||||
| Glc 1′′ | 4.96 | d | 7.5 | 101.7 | 4.95 | d | 7.5 | 102.5 | |
| 2′′ | 3.49 | dd | 8.5, 7.5 | 74.8 | 3.50 | dd | 8.7, 7.5 | 75.5 | |
| 3′′ | 3.46 | t | 8.5 | 78.1 | 3.46 | t | 8.7 | 78.8 | |
| 4′′ | 3.36 | dd | 9.2, 8.5 | 71.4 | 3.63 | t | 8.7 | 72.2 | |
| 5′′ | 3.59 | m | 77.3 | 3.59 | m | 78.1 | |||
| 6′′ | 4.02 | dd | 12.5, 5.0 | 67.6 | 4.01 | br d | 11.2 | 68.3 | |
| 3.59 | br d | 12.5 | 3.60 | br d | 11.0 | ||||
| Rha 1′′′ | 4.68 | br s | 102.2 | 4.68 | br s | 103.0 | |||
| 2′′′ | 3.88 | br s | 72.1 | 3.88 | br dd | 3.4, 1.4 | 72.9 | ||
| 3′′′ | 3.69 | dd | 9.6, 3.5 | 72.5 | 3.69 | dd | 9.8, 3.5 | 73.2 | |
| 4′′′ | 3.32 | t | 9.6 | 74.1 | 3.33 | t | 9.8 | 74.9 | |
| 5′′′ | 3.62 | m | 69.8 | 3.62 | m | 70.6 | |||
| 6′′′ | 1.18 | d | 6.3 | 18.0 | 1.18 | br d | 6.2 | 15.2 | |
| Compounds | IC50 (mM) |
|---|---|
| Cetilistat | 1.43 ± 0.052 μM |
| 1 | – |
| 2 | 0.84 ± 0.029 |
| 3 | 0.59 ± 0.020 |
| 4 | 1.42 ± 0.040 |
| 5 | 1.40 ± 0.035 |
| 6 | 0.81 ± 0.029 |
| 7 | 1.10 ± 0.015 |
| 8 | - |
| 9 | 0.91 ± 0.023 |
| 10 | 1.13 ± 0.062 |
| 11 | >4.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, Y.; Yamashiro, A.; Ootomo, K.; Nakagawa, M.; Tsuchihashi, H.; Inaba, N.; Mimaki, Y. Chemical Constituents of the Bulbs of Scilla peruviana and Their Pancreatic Lipase Inhibitory Activity. Int. J. Mol. Sci. 2021, 22, 1262. https://doi.org/10.3390/ijms22031262
Matsuo Y, Yamashiro A, Ootomo K, Nakagawa M, Tsuchihashi H, Inaba N, Mimaki Y. Chemical Constituents of the Bulbs of Scilla peruviana and Their Pancreatic Lipase Inhibitory Activity. International Journal of Molecular Sciences. 2021; 22(3):1262. https://doi.org/10.3390/ijms22031262
Chicago/Turabian StyleMatsuo, Yukiko, Asuka Yamashiro, Kanae Ootomo, Mika Nakagawa, Hiroko Tsuchihashi, Niro Inaba, and Yoshihiro Mimaki. 2021. "Chemical Constituents of the Bulbs of Scilla peruviana and Their Pancreatic Lipase Inhibitory Activity" International Journal of Molecular Sciences 22, no. 3: 1262. https://doi.org/10.3390/ijms22031262
APA StyleMatsuo, Y., Yamashiro, A., Ootomo, K., Nakagawa, M., Tsuchihashi, H., Inaba, N., & Mimaki, Y. (2021). Chemical Constituents of the Bulbs of Scilla peruviana and Their Pancreatic Lipase Inhibitory Activity. International Journal of Molecular Sciences, 22(3), 1262. https://doi.org/10.3390/ijms22031262