
Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
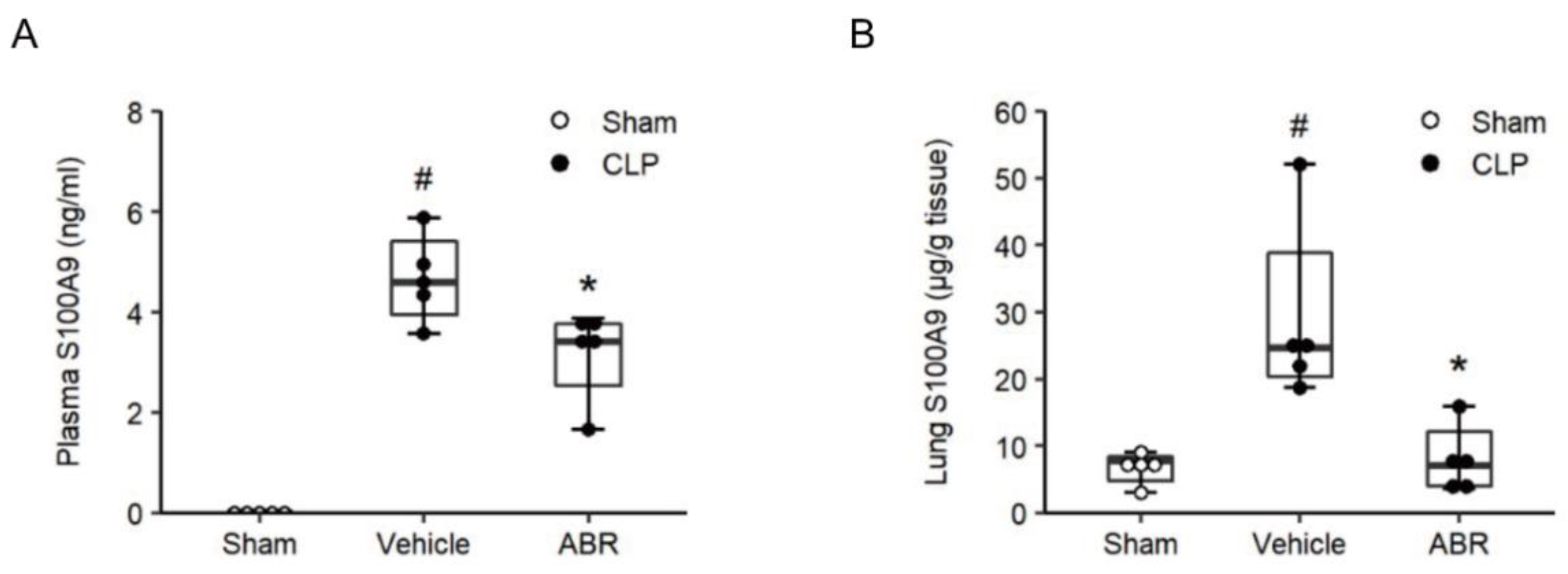
2.1. Septic Levels of S100A9
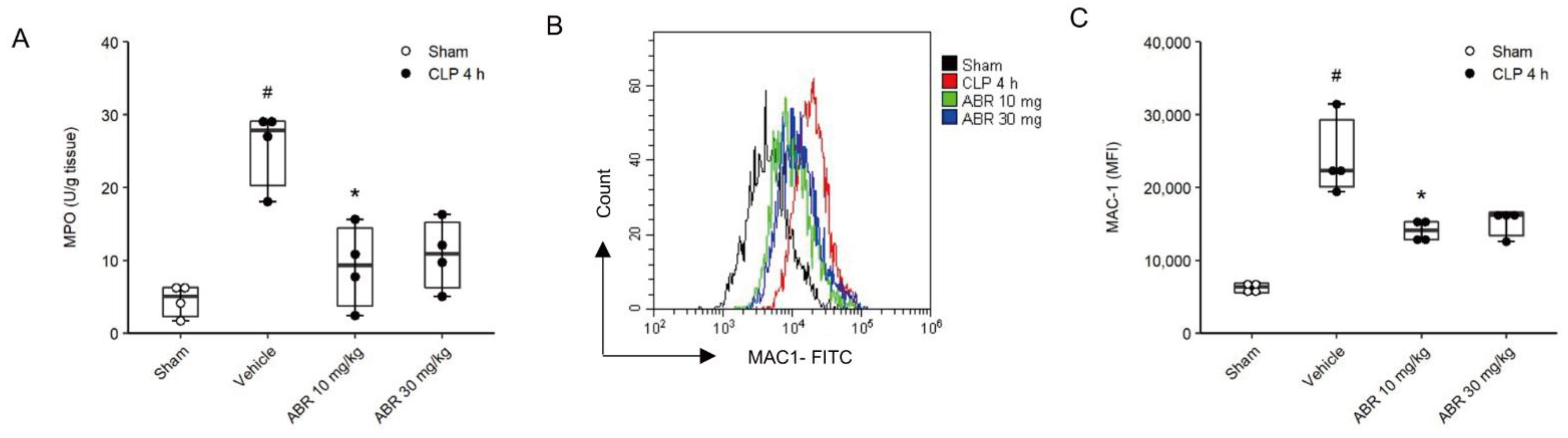
2.2. Neutrophil Recruitment and Mac-1 Expression
2.3. CXC Chemokines and IL-6 in the Lung
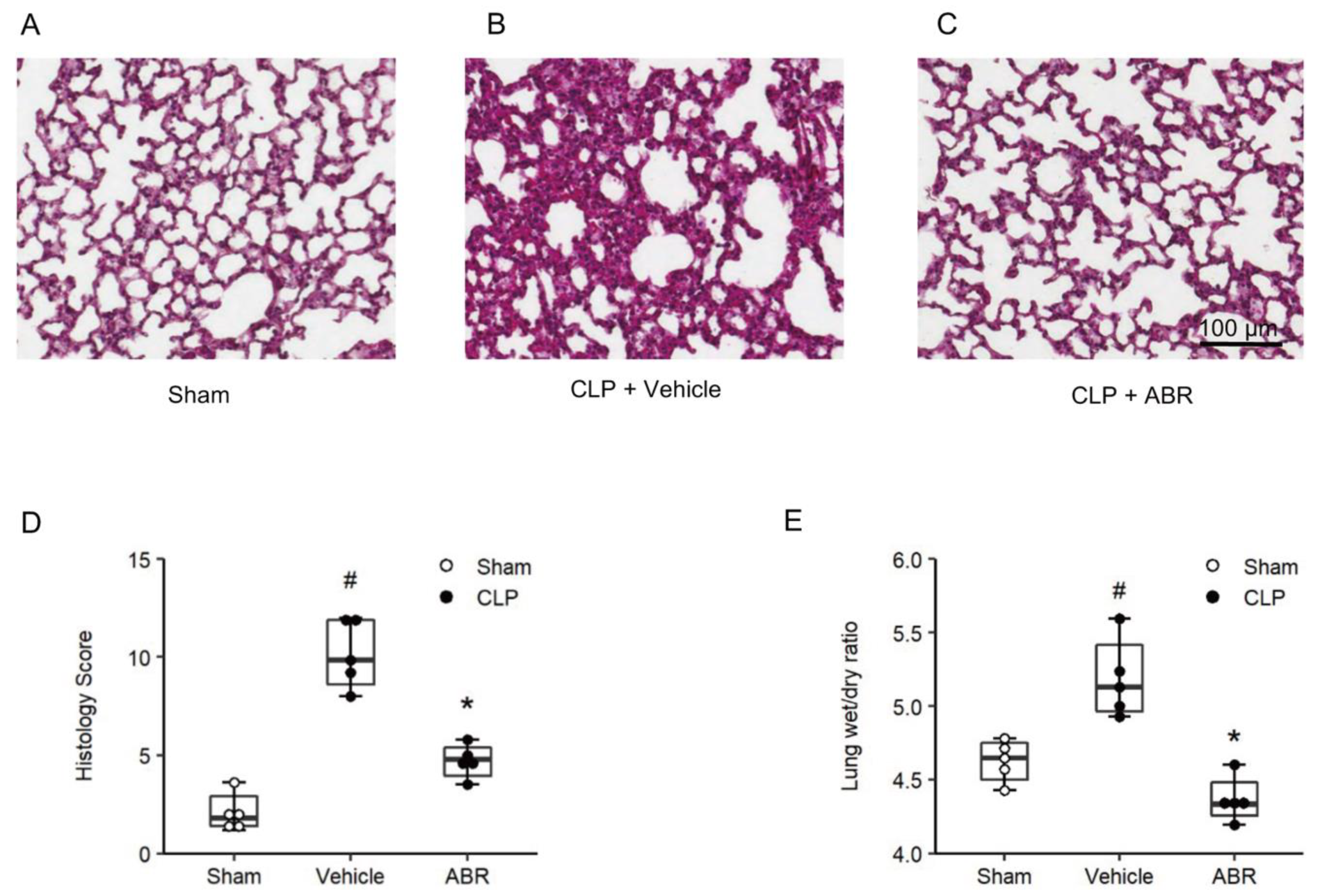
2.4. Lung Edema and Tissue Injury
2.5. Systemic Inflammation
2.6. Late Treatment with ABR-238901
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of CLP and Experimental Design
4.3. Myeloperoxidase Activity
4.4. Enzyme-Linked Immunosorbent Assay
4.5. Histology
4.6. Bronchoalveolar Lavage Fluid (BALF) and Lung Edema
4.7. Systemic Leukocytes Differential Counts
4.8. Flow Cytometry
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parrillo, J.E. Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 1993, 328, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Zhang, S.; Lavasani, S.; Wang, Y.; Thorlacius, H. LFA-1 and MAC-1 mediate pulmonary recruitment of neutrophils and tissue damage in abdominal sepsis. Shock 2008, 30, 254–259. [Google Scholar] [CrossRef]
- Zhang, S.; Rahman, M.; Zhang, S.; Qi, Z.; Thorlacius, H. Simvastatin antagonizes CD40L secretion, CXC chemokine formation, and pulmonary infiltration of neutrophils in abdominal sepsis. J. Leukoc. Biol. 2011, 89, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Z.; Palani, K.; Rahman, M.; Zhang, S.; Syk, I.; Jeppsson, B.; Thorlacius, H. Rho-kinase signaling regulates pulmonary infiltration of neutrophils in abdominal sepsis via attenuation of CXC chemokine formation and Mac-1 expression on neutrophils. Shock 2012, 37, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Hwaiz, R.; Hasan, Z.; Rahman, M.; Zhang, S.; Palani, K.; Syk, I.; Jeppsson, B.; Thorlacius, H. Rac1 signaling regulates sepsis-induced pathologic inflammation in the lung via attenuation of Mac-1 expression and CXC chemokine formation. J. Surg. Res. 2013, 183, 798–807. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, S.; Wang, Y.; Rahman, M.; Syk, I.; Zhang, E.; Thorlacius, H. Proinflammatory role of neutrophil extracellular traps in abdominal sepsis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L586–L596. [Google Scholar] [CrossRef]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef]
- Vogl, T.; Stratis, A.; Wixler, V.; Voller, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Frohling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Investig. 2018, 128, 1852–1866. [Google Scholar] [CrossRef]
- Edgeworth, J.; Gorman, M.; Bennett, R.; Freemont, P.; Hogg, N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J. Biol. Chem. 1991, 266, 7706–7713. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Zwadlo, G.; Bruggen, J.; Gerhards, G.; Schlegel, R.; Sorg, C. Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin. Exp. Immunol. 1988, 72, 510–515. [Google Scholar] [PubMed]
- Kumar, A.; Steinkasserer, A.; Berchtold, S. Interleukin-10 influences the expression of MRP8 and MRP14 in human dendritic cells. Int. Arch. Allergy Immunol. 2003, 132, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Warmlander, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, D.; Tenbrock, K.; Roth, J. Alarmins of the S100-Family in Juvenile Autoimmune and Auto-Inflammatory Diseases. Front. Immunol. 2019, 10, 182. [Google Scholar] [CrossRef]
- Vogl, T.; Eisenblatter, M.; Voller, T.; Zenker, S.; Hermann, S.; van Lent, P.; Faust, A.; Geyer, C.; Petersen, B.; Roebrock, K.; et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat. Commun. 2014, 5, 4593. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Marce, D.; Faivre, V.; Lukaszewicz, A.C.; Junot, C.; Fenaille, F.; Simon, S.; Becher, F.; Morel, N.; Payen, D. High plasma level of S100A8/S100A9 and S100A12 at admission indicates a higher risk of death in septic shock patients. Sci. Rep. 2019, 9, 15660. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Vandal, K.; Rouleau, P.; Boivin, A.; Ryckman, C.; Talbot, M.; Tessier, P.A. Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J. Immunol. 2003, 171, 2602–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raquil, M.A.; Anceriz, N.; Rouleau, P.; Tessier, P.A. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J. Immunol. 2008, 180, 3366–3374. [Google Scholar] [CrossRef] [Green Version]
- Lugering, N.; Stoll, R.; Kucharzik, T.; Schmid, K.W.; Rohlmann, G.; Burmeister, G.; Sorg, C.; Domschke, W. Immunohistochemical distribution and serum levels of the Ca(2+)-binding proteins MRP8, MRP14 and their heterodimeric form MRP8/14 in Crohn’s disease. Digestion 1995, 56, 406–414. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.; Wang, J.; Qin, Z.; Wang, J.; Lu, Y.; Zheng, X.; Peng, Q.; Ye, Q.; Ai, F.; et al. Suppression Colitis and Colitis-Associated Colon Cancer by Anti-S100a9 Antibody in Mice. Front. Immunol. 2017, 8, 1774. [Google Scholar] [CrossRef] [Green Version]
- Cesaro, A.; Anceriz, N.; Plante, A.; Page, N.; Tardif, M.R.; Tessier, P.A. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS ONE 2012, 7, e45478. [Google Scholar] [CrossRef]
- Marinkovic, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Zhao, B.; Lu, R.; Chen, J.; Xie, M.; Zhao, X.; Kong, L. S100A9 blockade prevents lipopolysaccharide-induced lung injury via suppressing the NLRP3 pathway. Respir. Res. 2021, 22, 45. [Google Scholar] [CrossRef]
- Xiang, H.; Guo, F.; Tao, X.; Zhou, Q.; Xia, S.; Deng, D.; Li, L.; Shang, D. Pancreatic ductal deletion of S100A9 alleviates acute pancreatitis by targeting VNN1-mediated ROS release to inhibit NLRP3 activation. Theranostics 2021, 11, 4467–4482. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Hsu, K.; Tedla, N.; Wong, S.W.; Chow, S.; Kawaguchi, N.; Geczy, C.L. S100A8/A9 and S100A9 reduce acute lung injury. Immunol. Cell Biol. 2017, 95, 461–472. [Google Scholar] [CrossRef]
- Van Zoelen, M.A.; Vogl, T.; Foell, D.; Van Veen, S.Q.; van Till, J.W.; Florquin, S.; Tanck, M.W.; Wittebole, X.; Laterre, P.F.; Boermeester, M.A.; et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am. J. Respir. Crit. Care Med. 2009, 180, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remick, D.G. Pathophysiology of sepsis. Am. J. Pathol. 2007, 170, 1435–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, Y.; Ode, Y.; Ochani, M.; Wang, P.; Aziz, M. Targeting junctional adhesion molecule-C ameliorates sepsis-induced acute lung injury by decreasing CXCR4(+) aged neutrophils. J. Leukoc. Biol. 2018, 104, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roller, J.; Slotta, J.E.; Zhang, S.; Luo, L.; Rahman, M.; Syk, I.; Menger, M.D.; Thorlacius, H. Distinct patterns of leukocyte recruitment in the pulmonary microvasculature in response to local and systemic inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L298–L305. [Google Scholar] [CrossRef]
- Wang, Y.; Roller, J.; Menger, M.D.; Thorlacius, H. Sepsis-induced leukocyte adhesion in the pulmonary microvasculature in vivo is mediated by CD11a and CD11b. Eur. J. Pharmacol. 2013, 702, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roller, J.; Wang, Y.; Rahman, M.; Schramm, R.; Laschke, M.W.; Menger, M.D.; Jeppsson, B.; Thorlacius, H. Direct in vivo observations of P-selectin glycoprotein ligand-1-mediated leukocyte-endothelial cell interactions in the pulmonary microvasculature in abdominal sepsis in mice. Inflamm. Res. 2013, 62, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P.A. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Bacon, K.B.; Oppenheim, J.J. Chemokines in disease models and pathogenesis. Cytokine Growth Factor Rev. 1998, 9, 167–173. [Google Scholar] [CrossRef]
- Ono, S.J.; Nakamura, T.; Miyazaki, D.; Ohbayashi, M.; Dawson, M.; Toda, M. Chemokines: Roles in leukocyte development, trafficking, and effector function. J. Allergy Clin. Immunol. 2003, 111, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Huang, S.; Danaee, H.; Paulauskis, J.D.; Kobzik, L. Functional characterization of the rat chemokine KC and its importance in neutrophil recruitment in a rat model of pulmonary inflammation. J. Immunol. 1995, 154, 335–344. [Google Scholar] [PubMed]
- Bhatia, M.; Zemans, R.L.; Jeyaseelan, S. Role of chemokines in the pathogenesis of acute lung injury. Am. J. Respir. Cell. Mol. Biol. 2012, 46, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukui, T.; Ehama, R.; Sakaguchi, M.; Sonegawa, H.; Katagiri, C.; Hibino, T.; Huh, N.H. S100A8/A9, a key mediator for positive feedback growth stimulation of normal human keratinocytes. J. Cell. Biochem. 2008, 104, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Wiechert, L.; Nemeth, J.; Pusterla, T.; Bauer, C.; De Ponti, A.; Manthey, S.; Marhenke, S.; Vogel, A.; Klingmuller, U.; Hess, J.; et al. Hepatocyte-specific S100a8 and S100a9 transgene expression in mice causes Cxcl1 induction and systemic neutrophil enrichment. Cell Commun. Signal. 2012, 10, 40. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PMNL | MNL | Total Leukocytes | |
|---|---|---|---|
| Sham | 1.2 ± 0.2 | 5.3 ± 1.9 | 6.5 ± 1.8 |
| Vehicle + CLP | 0.6 ± 0.3 # | 2.0 ± 0.8 # | 2.6 ± 1.1 # |
| ABR + CLP | 1.3 ± 0.5 * | 3.2 ± 0.7 | 4.6 ± 0.9 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Z.; Du, F.; Averitt V, R.G.; Jakobsson, G.; Rönnow, C.-F.; Rahman, M.; Schiopu, A.; Thorlacius, H. Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. Int. J. Mol. Sci. 2021, 22, 12923. https://doi.org/10.3390/ijms222312923
Ding Z, Du F, Averitt V RG, Jakobsson G, Rönnow C-F, Rahman M, Schiopu A, Thorlacius H. Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. International Journal of Molecular Sciences. 2021; 22(23):12923. https://doi.org/10.3390/ijms222312923
Chicago/Turabian StyleDing, Zhiyi, Feifei Du, Richard Garland Averitt V, Gabriel Jakobsson, Carl-Fredrik Rönnow, Milladur Rahman, Alexandru Schiopu, and Henrik Thorlacius. 2021. "Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis" International Journal of Molecular Sciences 22, no. 23: 12923. https://doi.org/10.3390/ijms222312923
APA StyleDing, Z., Du, F., Averitt V, R. G., Jakobsson, G., Rönnow, C.-F., Rahman, M., Schiopu, A., & Thorlacius, H. (2021). Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. International Journal of Molecular Sciences, 22(23), 12923. https://doi.org/10.3390/ijms222312923