
CRB1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Müller Cell and Photoreceptor CRB1 Isoforms

 , , , ,
, , , ,  , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
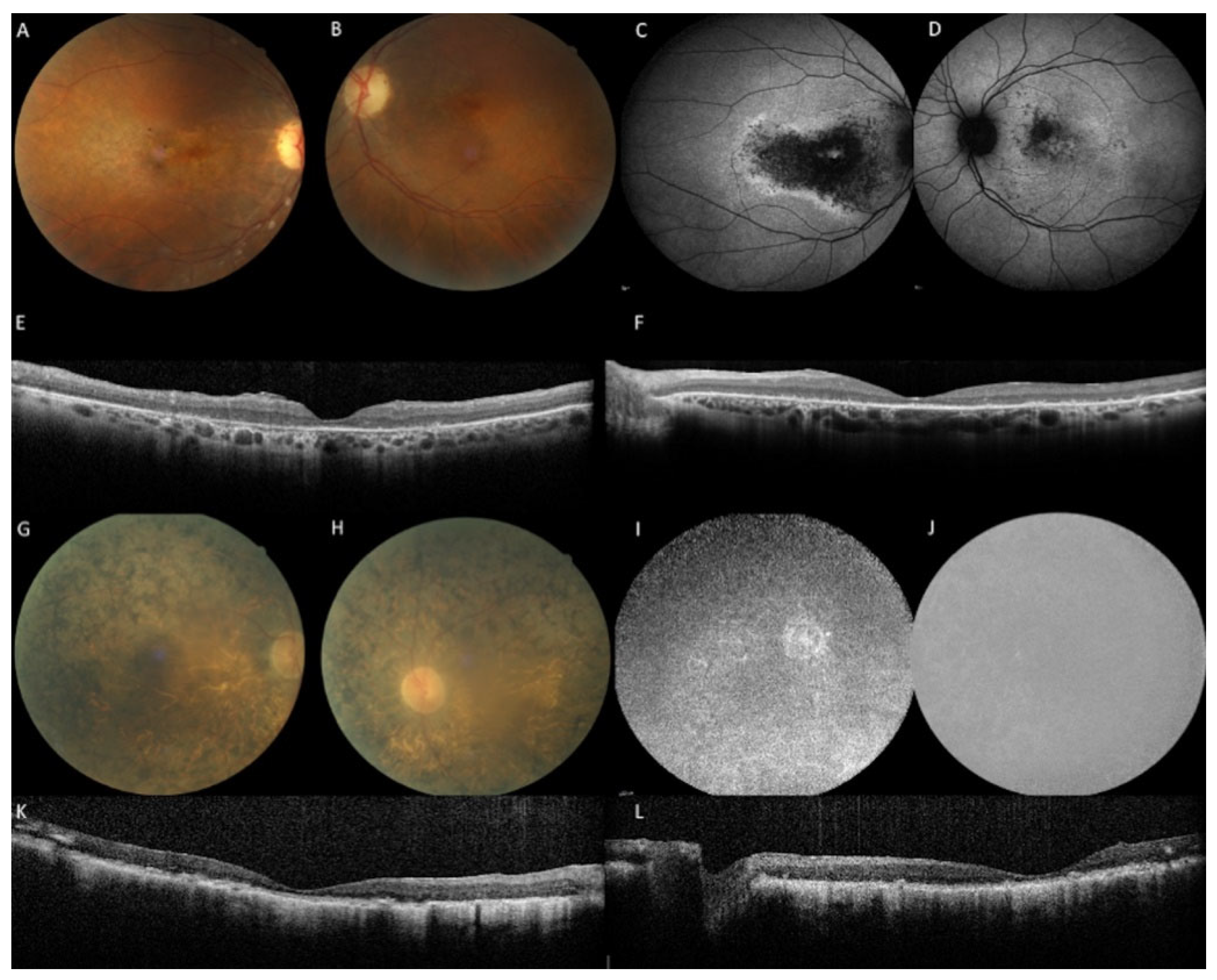
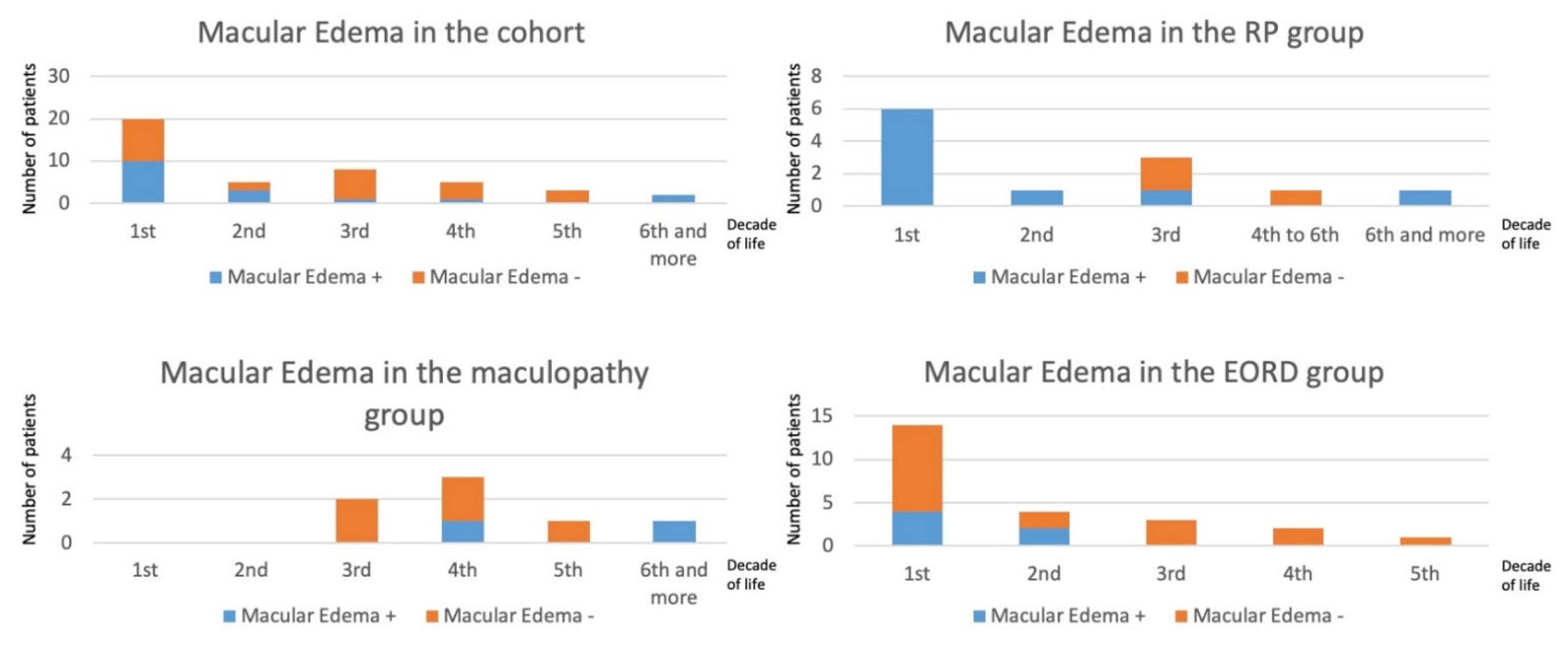
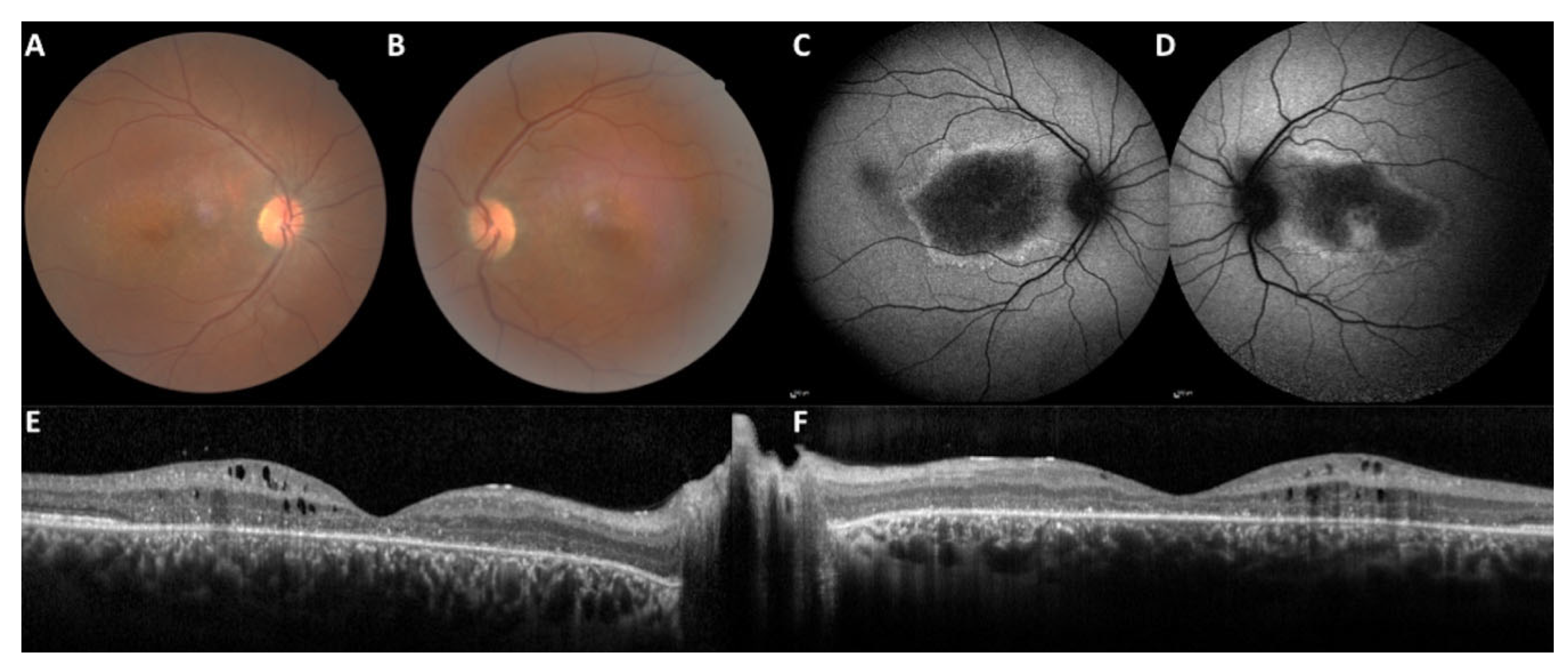
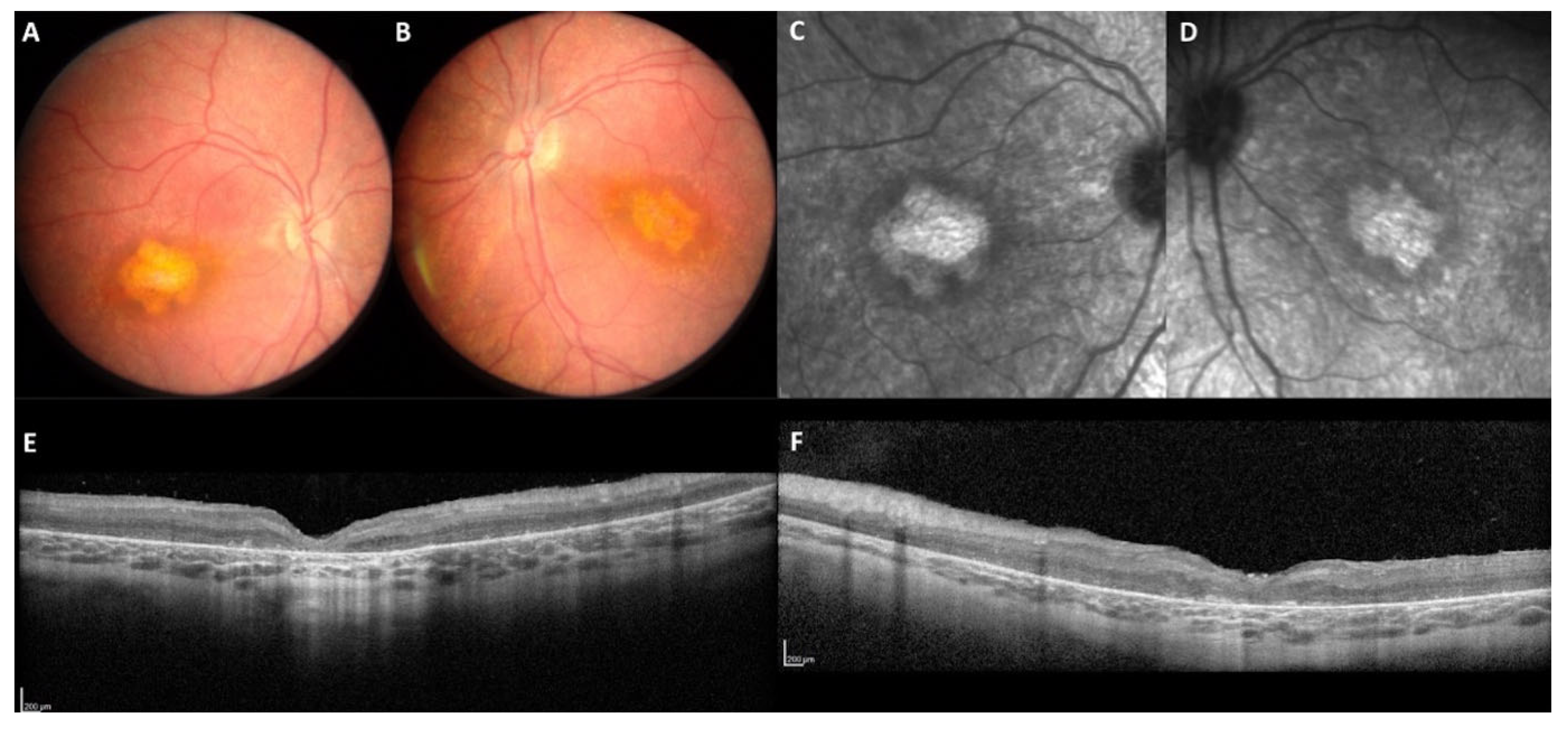
2.1. Clinical Data
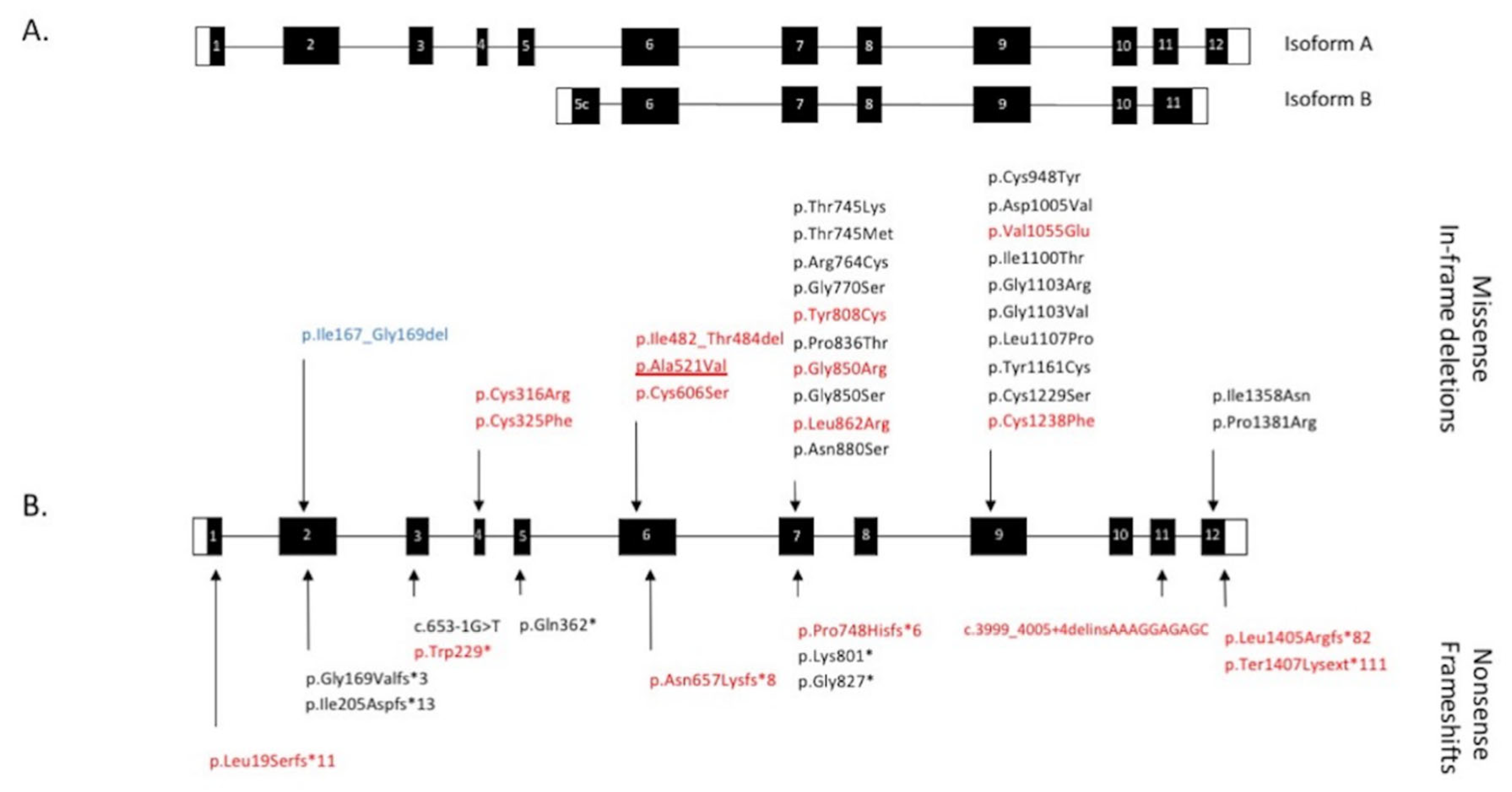
2.2. Genetic Results
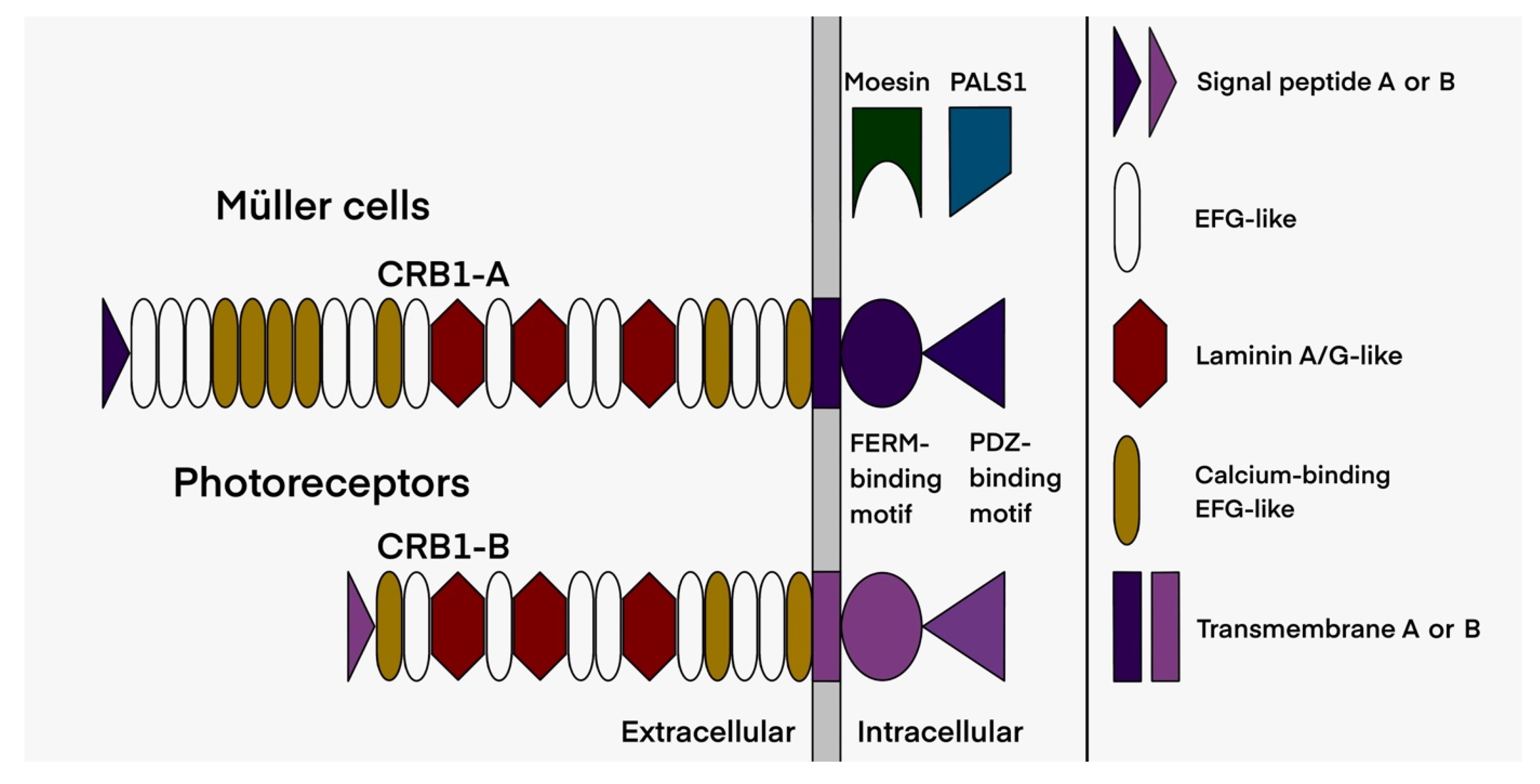
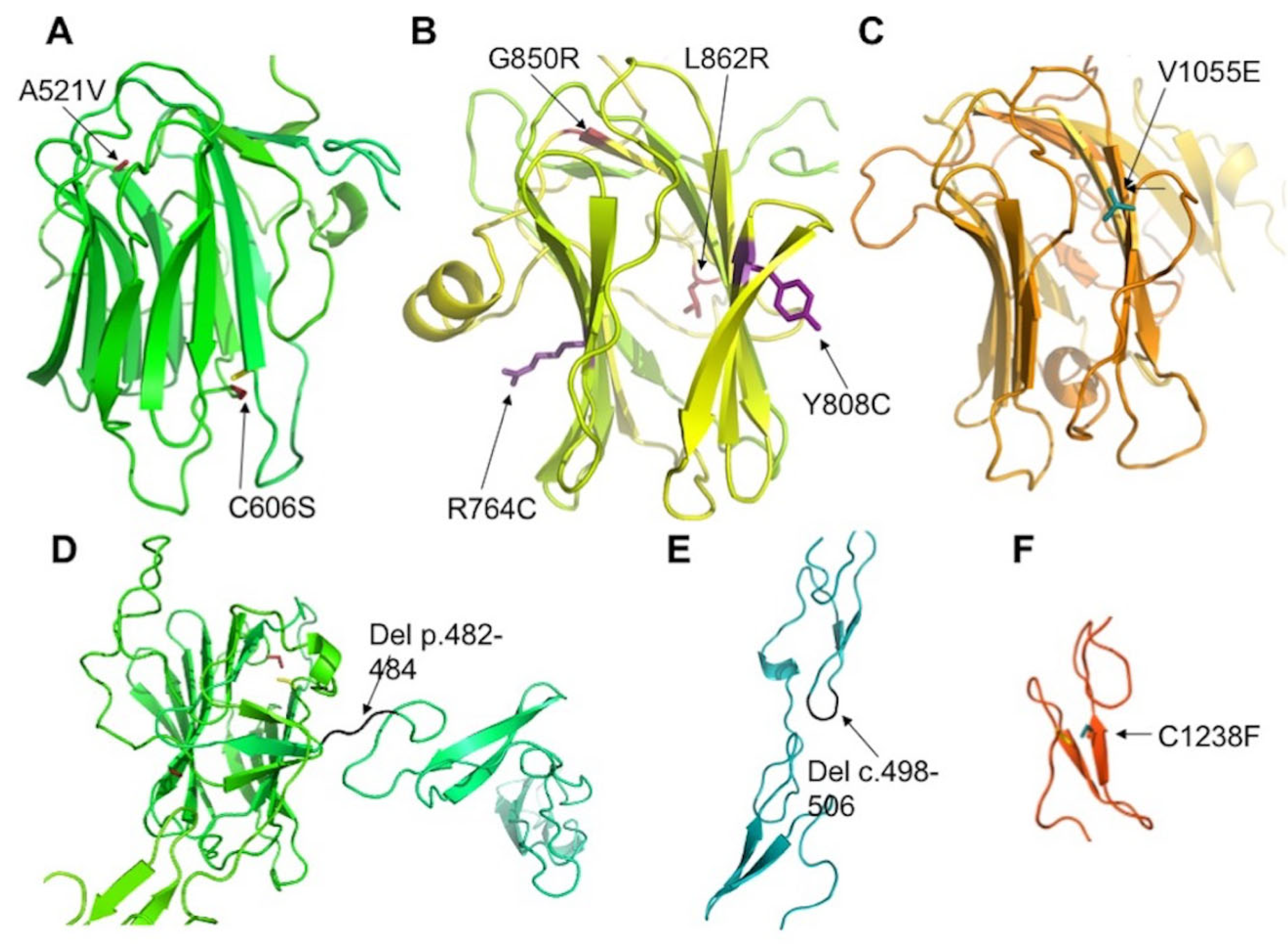
2.3. Assessment of the Functional Effect of CRB1 Variants
2.4. Impact of CRB1-A and CRB1-B Variants on the Phenotype
3. Discussion
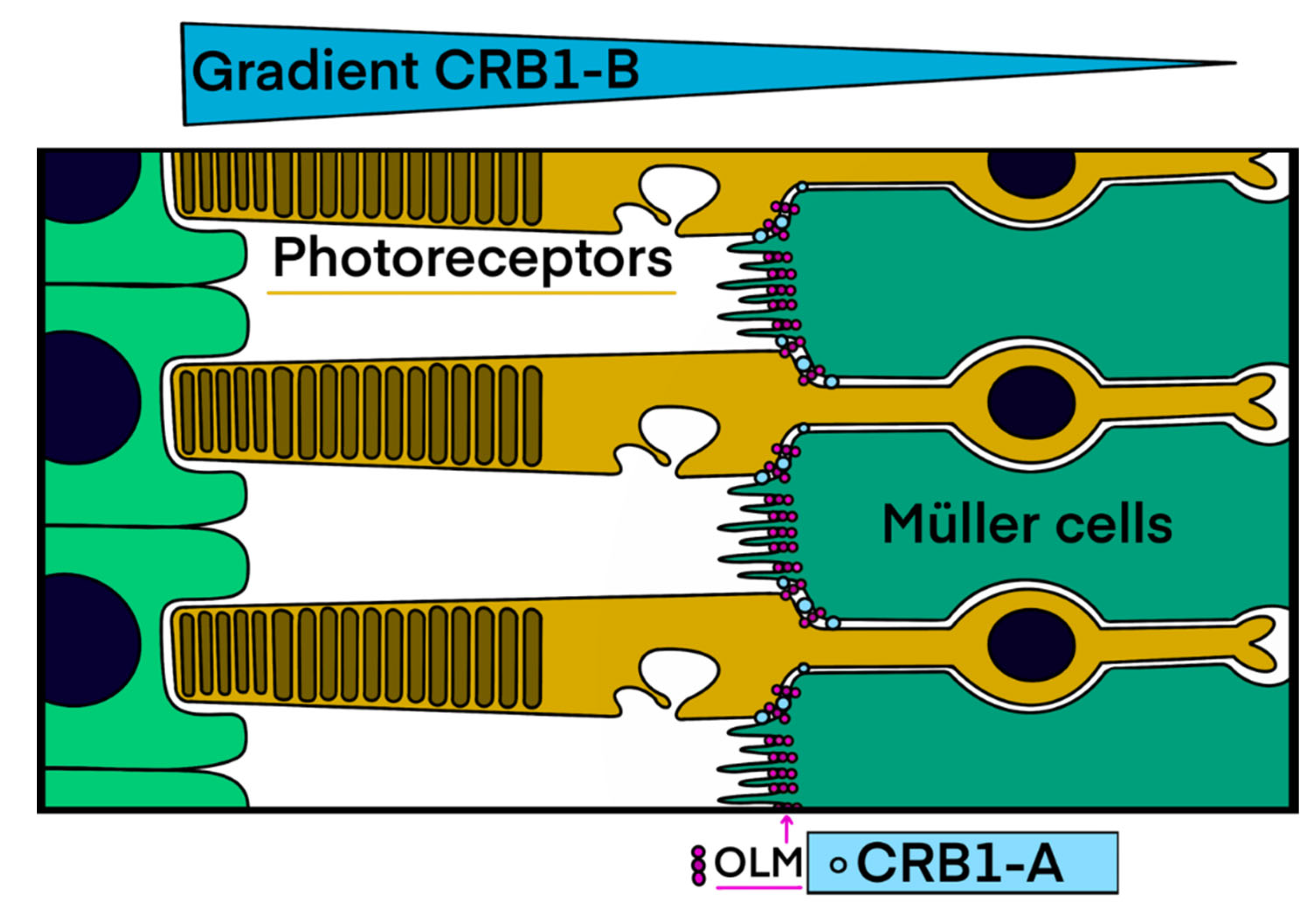
3.1. CRB1-Related Retinal Dystrophies
3.1.1. The Macular Phenotype Is Determined by the c.498_506del Variant Specific to CRB1-A
3.1.2. CRB1 Variants and Structure Function Correlation in EORD and in RP
3.2. Intrafamilial Variation Is an Exception
3.3. Limitations of Mouse Models, Concordance and Discordance
3.4. Future Directions
4. Materials and Methods
4.1. Clinical Investigation
4.2. SD-OCT Measurements
4.3. Molecular Investigation
4.4. Variant Analysis and Protein Modelling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRB1 | Crumbs homologue 1 |
| EORD | Early onset retinal dystrophy |
| IRD | Inherited retinal dystrophy |
| LCA | Leber congenital amaurosis |
| MD RP, RCD | Macular dystrophy Retinitis pigmentosa, rod cone dystrophy |
| SD-OCT | Spectral domain optical coherence tomography |
| EORD | Early onset retinal dystrophy |
References
- Hamel, C.P. Cone Rod Dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Bocquet, B.; Lacroux, A.; Surget, M.-O.; Baudoin, C.; Marquette, V.; Manes, G.; Hebrard, M.; Sénéchal, A.; Delettre, C.; Roux, A.-F.; et al. Relative Frequencies of Inherited Retinal Dystrophies and Optic Neuropathies in Southern France: Assessment of 21-Year Data Management. Ophthalmic Epidemiol. 2013, 20, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahim, A.T.; Bouzia, Z.; Branham, K.H.; Kumaran, N.; Vargas, M.E.; Feathers, K.L.; Perera, N.D.; Young, K.; Khan, N.W.; Heckenlively, J.R.; et al. Detailed Clinical Characterisation, Unique Features and Natural History of Autosomal Recessive RDH12-Associated Retinal Degeneration. Br. J. Ophthalmol. 2019, 103, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.A.; Place, E.M.; Ferenchak, K.; Zampaglione, E.; Wagner, N.E.; Chao, K.R.; DiTroia, S.P.; Navarro-Gomez, D.; Mukai, S.; Huckfeldt, R.M.; et al. Expanding the Phenotypic Spectrum in RDH12-Associated Retinal Disease. Cold Spring Harb. Mol. Case Stud. 2020, 6, a004754. [Google Scholar] [CrossRef] [Green Version]
- Hull, S.; Arno, G.; Plagnol, V.; Chamney, S.; Russell-Eggitt, I.; Thompson, D.; Ramsden, S.C.; Black, G.C.M.; Robson, A.; Holder, G.E.; et al. The Phenotypic Variability of Retinal Dystrophies Associated with Mutations in CRX, with Report of a Novel Macular Dystrophy Phenotype. Invest Ophthalmol. Vis. Sci. 2014, 55, 6934–6944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and Genetic Characteristics of 251 Consecutive Patients with Macular and Cone/Cone-Rod Dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Burke, T.; Oll, M.; Yzer, S.; Lee, W.; Xie, Y.; Allikmets, R. Whole Exome Sequencing Identifies CRB1 Defect in an Unusual Maculopathy Phenotype. Ophthalmology 2014, 121, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Zhang, Q. Clinical and Genetic Analysis of 63 Families Demonstrating Early and Advanced Characteristic Fundus as the Signature of CRB1 Mutations. Am. J. Ophthalmol. 2021, 223, 160–168. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Davis, J.; van der Velde-Visser, S.D.; Zonneveld, M.N.; Pierrottet, C.O.; Koenekoop, R.K.; Kellner, U.; van den Born, L.I.; Heckenlively, J.R.; Hoyng, C.B.; et al. CRB1 Mutation Spectrum in Inherited Retinal Dystrophies. Hum. Mutat 2004, 24, 355–369. [Google Scholar] [CrossRef]
- Bujakowska, K.; Audo, I.; Mohand-Saïd, S.; Lancelot, M.-E.; Antonio, A.; Germain, A.; Léveillard, T.; Letexier, M.; Saraiva, J.-P.; Lonjou, C.; et al. CRB1 Mutations in Inherited Retinal Dystrophies. Hum. Mutat. 2012, 33, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Lotery, A.J.; Malik, A.; Shami, S.A.; Sindhi, M.; Chohan, B.; Maqbool, C.; Moore, P.A.; Denton, M.J.; Stone, E.M. CRB1 Mutations May Result in Retinitis Pigmentosa without Para-Arteriolar RPE Preservation. Ophthalmic Genet. 2001, 22, 163–169. [Google Scholar] [CrossRef]
- Nguyen, X.-T.-A.; Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Schalij-Delfos, N.E.; Klaver, C.C.W.; Talsma, H.E.; Fiocco, M.; Florijn, R.J.; et al. CRB1-Associated Retinal Dystrophies: A Prospective Natural History Study in Anticipation of Future Clinical Trials. Am. J. Ophthalmol. 2021, 234, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs Homolog 1 (CRB1) Mutations Result in a Thick Human Retina with Abnormal Lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulou Laiou, C.; Preising, M.N.; Bolz, H.J.; Lorenz, B. [Genotype-Phenotype Correlations in Patients with CRB1 Mutations]. Klin. Monbl. Augenheilkd 2017, 234, 289–302. [Google Scholar] [PubMed]
- Talib, M.; van Schooneveld, M.J.; van Genderen, M.M.; Wijnholds, J.; Florijn, R.J.; Ten Brink, J.B.; Schalij-Delfos, N.E.; Dagnelie, G.; Cremers, F.P.M.; Wolterbeek, R.; et al. Genotypic and Phenotypic Characteristics of CRB1-Associated Retinal Dystrophies: A Long-Term Follow-up Study. Ophthalmology 2017, 124, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Corton, M.; Tatu, S.D.; Avila-Fernandez, A.; Vallespín, E.; Tapias, I.; Cantalapiedra, D.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Bernal, S.; García-Sandoval, B.; et al. High Frequency of CRB1 Mutations as Cause of Early-Onset Retinal Dystrophies in the Spanish Population. Orphanet J. Rare Dis. 2013, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Alves, C.H.; Pellissier, L.P.; Wijnholds, J. The CRB1 and Adherens Junction Complex Proteins in Retinal Development and Maintenance. Prog. Retin Eye Res. 2014, 40, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Mehalow, A.K.; Kameya, S.; Smith, R.S.; Hawes, N.L.; Denegre, J.M.; Young, J.A.; Bechtold, L.; Haider, N.B.; Tepass, U.; Heckenlively, J.R.; et al. CRB1 Is Essential for External Limiting Membrane Integrity and Photoreceptor Morphogenesis in the Mammalian Retina. Hum. Mol. Genet. 2003, 12, 2179–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Pavert, S.A.; Kantardzhieva, A.; Malysheva, A.; Meuleman, J.; Versteeg, I.; Levelt, C.; Klooster, J.; Geiger, S.; Seeliger, M.W.; Rashbass, P.; et al. Crumbs Homologue 1 Is Required for Maintenance of Photoreceptor Cell Polarization and Adhesion during Light Exposure. J. Cell Sci. 2004, 117, 4169–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Cho, M.; Lee, J. Crumbs Proteins Regulate Layered Retinal Vascular Development Required for Vision. Biochem Biophys Res. Commun. 2020, 521, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive Identification of MRNA Isoforms Reveals the Diversity of Neural Cell-Surface Molecules with Roles in Retinal Development and Disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]
- Khan, K.N.; Robson, A.; Mahroo, O.A.R.; Arno, G.; Inglehearn, C.F.; Armengol, M.; Waseem, N.; Holder, G.E.; Carss, K.J.; Raymond, L.F.; et al. A Clinical and Molecular Characterisation of CRB1-Associated Maculopathy. Eur. J. Hum. Genet. 2018, 26, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Hollander, A.I.; ten Brink, J.B.; de Kok, Y.J.M.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.R.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a Human Homologue of Drosophila Crumbs Cause Retinitis Pigmentosa (RP12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-Based next Generation Sequencing as a Reliable and Efficient Technique to Detect Mutations in Unselected Patients with Retinal Dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Lotery, A.J.; Jacobson, S.G.; Fishman, G.A.; Weleber, R.G.; Fulton, A.B.; Namperumalsamy, P.; Héon, E.; Levin, A.V.; Grover, S.; Rosenow, J.R.; et al. Mutations in the CRB1 Gene Cause Leber Congenital Amaurosis. Arch. Ophthalmol. 2001, 119, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Yzer, S.; Leroy, B.P.; De Baere, E.; de Ravel, T.J.; Zonneveld, M.N.; Voesenek, K.; Kellner, U.; Ciriano, J.P.M.; de Faber, J.-T.H.N.; Rohrschneider, K.; et al. Microarray-Based Mutation Detection and Phenotypic Characterization of Patients with Leber Congenital Amaurosis. Invest Ophthalmol. Vis. Sci. 2006, 47, 1167–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanks, M.E.; Downes, S.M.; Copley, R.R.; Lise, S.; Broxholme, J.; Hudspith, K.A.; Kwasniewska, A.; Davies, W.I.; Hankins, M.W.; Packham, E.R.; et al. Next-Generation Sequencing (NGS) as a Diagnostic Tool for Retinal Degeneration Reveals a Much Higher Detection Rate in Early-Onset Disease. Eur. J. Hum. Genet. 2013, 21, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Kousal, B.; Dudakova, L.; Gaillyova, R.; Hejtmankova, M.; Diblik, P.; Michaelides, M.; Liskova, P. Phenotypic Features of CRB1-Associated Early-Onset Severe Retinal Dystrophy and the Different Molecular Approaches to Identifying the Disease-Causing Variants. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 1833–1839. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Heckenlively, J.R.; van den Born, L.I.; de Kok, Y.J.; van der Velde-Visser, S.D.; Kellner, U.; Jurklies, B.; van Schooneveld, M.J.; Blankenagel, A.; Rohrschneider, K.; et al. Leber Congenital Amaurosis and Retinitis Pigmentosa with Coats-like Exudative Vasculopathy Are Associated with Mutations in the Crumbs Homologue 1 (CRB1) Gene. Am. J. Hum. Genet. 2001, 69, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanein, S.; Perrault, I.; Gerber, S.; Tanguy, G.; Barbet, F.; Ducroq, D.; Calvas, P.; Dollfus, H.; Hamel, C.; Lopponen, T.; et al. Leber Congenital Amaurosis: Comprehensive Survey of the Genetic Heterogeneity, Refinement of the Clinical Definition, and Genotype-Phenotype Correlations as a Strategy for Molecular Diagnosis. Hum. Mutat 2004, 23, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, S.; Calaf, M.; Garcia-Hoyos, M.; Garcia-Sandoval, B.; Rosell, J.; Adan, A.; Ayuso, C.; Baiget, M. Study of the Involvement of the RGR, CRPB1, and CRB1 Genes in the Pathogenesis of Autosomal Recessive Retinitis Pigmentosa. J. Med. Genet. 2003, 40, e89. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Külm, M.; Dharmaraj, S.; den Hollander, A.I.; Perrault, I.; Preising, M.N.; Lorenz, B.; Kaplan, J.; Cremers, F.P.M.; Maumenee, I.; et al. Genotyping Microarray (Disease Chip) for Leber Congenital Amaurosis: Detection of Modifier Alleles. Invest Ophthalmol. Vis. Sci. 2005, 46, 3052–3059. [Google Scholar] [CrossRef]
- Vallespin, E.; Avila-Fernandez, A.; Velez-Monsalve, C.; Almoguera, B.; Martinez-Garcia, M.; Gomez-Dominguez, B.; Gonzalez-Roubaud, C.; Cantalapiedra, D.; Trujillo-Tiebas, M.J.; Ayuso, C. Novel Human Pathological Mutations. Gene Symbol: CRB1. Disease: Leber Congenital Amaurosis. Hum. Genet. 2010, 127, 119. [Google Scholar] [PubMed]
- Zaneveld, J.; Siddiqui, S.; Li, H.; Wang, X.; Wang, H.; Wang, K.; Li, H.; Ren, H.; Lopez, I.; Dorfman, A.; et al. Comprehensive Analysis of Patients with Stargardt Macular Dystrophy Reveals New Genotype-Phenotype Correlations and Unexpected Diagnostic Revisions. Genet. Med. 2015, 17, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xiao, X.; Li, S.; Jia, X.; Xin, W.; Wang, P.; Sun, W.; Huang, L.; Guo, X.; Zhang, Q. Molecular Genetics of Leber Congenital Amaurosis in Chinese: New Data from 66 Probands and Mutation Overview of 159 Probands. Exp. Eye Res. 2016, 149, 93–99. [Google Scholar] [CrossRef]
- Henderson, R.H.; Mackay, D.S.; Li, Z.; Moradi, P.; Sergouniotis, P.; Russell-Eggitt, I.; Thompson, D.A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; et al. Phenotypic Variability in Patients with Retinal Dystrophies Due to Mutations in CRB1. Br. J. Ophthalmol. 2011, 95, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Kantardzhieva, A.; Gosens, I.; Alexeeva, S.; Punte, I.M.; Versteeg, I.; Krieger, E.; Neefjes-Mol, C.A.; den Hollander, A.I.; Letteboer, S.J.F.; Klooster, J.; et al. MPP5 Recruits MPP4 to the CRB1 Complex in Photoreceptors. Invest Ophthalmol. Vis. Sci. 2005, 46, 2192–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.; Alves, C.H.; Lundvig, D.M.; Tanimoto, N.; Beck, S.C.; Huber, G.; Richard, F.; Klooster, J.; Andlauer, T.F.M.; Swindell, E.C.; et al. PALS1 Is Essential for Retinal Pigment Epithelium Structure and Neural Retina Stratification. J. Neurosci. 2011, 31, 17230–17241. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, A.G.S.H.; Aartsen, W.M.; Meuleman, J.; Klooster, J.; Malysheva, A.; Versteeg, I.; Arsanto, J.-P.; Le Bivic, A.; Wijnholds, J. Pals1/Mpp5 Is Required for Correct Localization of Crb1 at the Subapical Region in Polarized Muller Glia Cells. Hum. Mol. Genet. 2006, 15, 2659–2672. [Google Scholar] [CrossRef]
- Wei, Z.; Li, Y.; Ye, F.; Zhang, M. Structural Basis for the Phosphorylation-Regulated Interaction between the Cytoplasmic Tail of Cell Polarity Protein Crumbs and the Actin-Binding Protein Moesin. J. Biol. Chem. 2015, 290, 11384–11392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañibano-Hernández, A.; Valdes-Sanchez, L.; Garcia-Delgado, A.B.; Ponte-Zúñiga, B.; Diaz-Corrales, F.J.; de la Cerda, B. Generation of the Human IPSC Line ESi082-A from a Patient with Macular Dystrophy Associated to Mutations in the CRB1 Gene. Stem Cell Res. 2021, 53, 102301. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.; Grunewald, O.; Muller, J.; Zeitz, C.; Obermaier, C.D.; Devos, A.; Pelletier, V.; Bocquet, B.; Andrieu, C.; Bacquet, J.-L.; et al. Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. Int. J. Mol. Sci. 2021, 22, 6410. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Canutescu, A.A.; Dunbrack, R.L. SCWRL and MolIDE: Computer Programs for Side-Chain Conformation Prediction and Homology Modeling. Nat. Protoc. 2008, 3, 1832–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number (Gender) | Variant 2 cDNA Protein | Age at Onset Age at First Visit | Refraction (SE) OD OG | Variant 1 cDNA Protein | Initial BCVA (logMAR, Snellen) OD OG | Final BCVA (logMAR, Snellen) OD OG | Follow up (Months) Age at Final Visit | Phenotype | Optic Disc Drusen | PPARPE | Coats-Like | Macular Oedema |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M-1640-1 | c.1562C>T | 3 | 2.25 | c.1562C>T | 2.3 (HM) | 2.3 (HM) | 82 | EORD | No | No | No | No |
| (M) | p.Ala521Val | 46.5 | 2 | p.Ala521Val | 2.3 (HM) | 2.6 (LP) | 53 | |||||
| M-2129 | c.2234C>T | 1.5 | 3.25 | c.2234C>T | 0.3 (20/40) | 0.8 (20/125) | 66 | EORD | No | Yes | No | No |
| (M) | p.Thr745Met | 9.5 | 3.25 | p.Thr745Met | 1.0 (20/200) | 0.8 (20/125) | 15 | |||||
| M-567 | c.1445_1453del | 3 | −0.25 | c.2290C>T | 0 (20/20) | 0.7 (20/100) | 84 | EORD | Yes | No | No | Yes |
| (M) | p.Ile482_Thr484del | 4 | −0.50 | p.Arg764Cys | 0 (20/20) | 1.3 (20/400) | 11 | |||||
| M-116 | c.4219T>A | 5 | 2 | c.687G>A | 0.7 (20/100) | ND | ND | EORD | ND | ND | ND | ND |
| (F) | p.Ter1407Lysext*111 | 43.5 | 2 | p.Trp229* | 0.7 (20/100) | |||||||
| M-3361 | c.2290C>T | 4.5 | 2.5 | c.2290C>T | 0.1 (20/25) | 0.3 (20/40) | 25 | EORD | Yes | No | No | Yes |
| (M) | p.Arg764Cys | 5 | 3.5 | p.Arg764Cys | 0.4 (20/50) | 0.3 (20/40) | 7.5 | |||||
| M-3591 | c.2401A>T | 1 | 2.75 | c.2290C>T | 0.7 (20/100) | 0.3 (20/40) | 23 | EORD | No | Yes | No | Yes |
| (M) | p.Lys801* | 4.5 | 3.25 | p.Arg764Cys | 0.7 (20/100) | 0.3 (20/40) | 6 | |||||
| M-2897 | c.2843G>A | 5 | 6.25 | c.2548G>A | 2.3 (HM) | ND | ND | EORD | No | No | No | No |
| (F) | p.Cys948Tyr | 34 | 6 | p.Gly850Ser | 1.7 (20/1000) | |||||||
| M-3235 | c.4214del | 4 | 0.5 | c.2290C>T | 0.7 (20/100) | ND | ND | EORD | Yes | Yes | No | Yes |
| (M) | p.Leu1405Argfs*82 | 17.5 | 1.75 | p.Arg764Cys | 0.7 (20/100) | |||||||
| M-1316-4 | c.2243del | 5 | 3.75 | c.2243del | 0.4 (20/50) | ND | ND | EORD | No | Yes | No | No |
| (F) | p.Pro748Hisfs*6 | 7 | 1.75 | p.Pro748Hisfs*6 | 0.7 (20/100) | |||||||
| M-340 | c.2479G>T | 1 | 3.75 | c.2290C>T | 1.0 (20/200) | 2.3 (HM) | 200 | EORD | Yes | Yes | No | No |
| (F) | p.Gly827* | 5 | 3.25 | p.Arg764Cys | 1.0 (20/200) | 1.3 (20/400) | 22 | |||||
| L- 15090708 (M) | c.3713G>T | 1 | 10 | c.2401A>T | 1.3 (20/400) | 1.1 (20/250) | 48 | EORD | No | Yes | No | No |
| p.Cys1238Phe | 4 | 10 | p.Lys801* | 1.0 (20/200) | 1.1 (20/250) | 8 | ||||||
| L-14061710 | c.4142C>G | 1 | 7 | c.2548G>A | 0.7 (20/100) | 0.4 (20/50) | 84 | EORD | Yes | No | No | No |
| (F) | p.Pro1381Arg | 4 | 7 | p.Gly850Ser | 0.7 (20/100) | 0.4 (20/50) | 11 | |||||
| XZ-372875 | c.3320T>C | 4 | 3 | c.3686G>C | 0.8 (20/125) | 0.7 (20/100) | 72 | EORD | No | Yes | No | No |
| (F) | p.Leu1107Pro | 4 | 4 | p.Cys1229Ser | 0.9 (20/160) | 0.7 (20/100) | 10 | |||||
| M-1316-1 | c.2243del | 3.5 | 3.75 | c.2243del | 1.2 (20/320) | 2.3 (HM) | 116 | EORD | No | Yes | No | No |
| (F) | p.Pro748Hisfs*67 | 4.5 | 1.75 | p.Pro748Hisfs*6 | 1.0 (20/200) | 1.3 (20/400) | 17 | |||||
| M-3324 | c.2401A>T | 1.5 | ND | c.1084C>T | 2.3 (HM) | ND | ND | EORD | No | No | No | No |
| (F) | p.Lys801* | 27 | ND | p.Gln362* | 2.3 (HM) | |||||||
| M-2427 | c.4073T>A | <5 | 7.5 | c.4073T>A | 2.3 (HM) | 2.0 (20/2000) | 26 | EORD | No | No | No | No |
| (M) | p.Ile1358Asn | 5 | 8 | p.Ile1358Asn | 2.3 (HM) | 2.0 (20/2000) | 7 | |||||
| M-4621-1 | c.4142C>G | 2.5 | 5.75 | c.4142C>G | ND | 0.8 (20/125) | 21 | EORD | No | Yes | No | No |
| (F) | p.Pro1381Arg | 3 | 6.5 | p.Pro1381Arg | ND | 0.8 (20/125) | 4.5 | |||||
| M-1580 | c.3320T>C | 1 | 4.5 | c.3164T>A | 2.3 (HM) | 2.6 (LP) | 47 | EORD + nystagmus | Yes | Yes | No | No |
| (F) | p.Leu1107Pro | 17 | 5.25 | p.Val1055Glu | 2.3 (HM) | 2.6 (LP) | 21 | |||||
| M-2123 | c.3999_4005+4delinsAAAGGAGAGC | 0.5 | ND | c.3999_4005+4delinsAAAGGAGAGC | ND | ND | ND | EORD + nystagmus | No | No | No | No |
| (M) | p.? | 0.5 | ND | p.? | ND | ND | ||||||
| M-2123-2 | c.3999_4005+4delinsAAAGGAGAGC | 0.4 | 4 | c.3999_4005+4delinsAAAGGAGAGC | ND | ND | ND | EORD + nystagmus | No | No | No | No |
| (M) | p.? | 0.5 | 4.5 | p.? | ND | ND | ||||||
| M-2186 | c.1971del | 0.5 | 3.5 | c.1971del | 2.3 (HM) | 2.3 (HM) | 76 | EORD + nystagmus | No | No | No | ND |
| (F) | p.Asn657Lysfs*8 | 1 | 5 | p.Asn657Lysfs*8 | 2.3 (HM) | 2.3 (HM) | 7 | |||||
| M-2184 | c.2843G>A | 4 | 3.75 | c.2401A>T | 1.0 (20/200) | 1.3 (20/400) | 82 | EORD + nystagmus | Yes | Yes | Yes | Yes |
| (F) | p.Cys948Tyr | 9 | 4.25 | p.Lys801* | 1.3 (20/400) | 1.3 (20/400) | 16 | |||||
| XZ-301265-1 (F) | c.2291G>A | 5 | 7.75 | c.2291G>A | 2.3 (HM) | 2.3 (HM) | 60 | EORD + nystagmus | No | No | No | No |
| p.Arg764His | 26 | 5.75 | p.Arg764His | 2.3 (HM) | 2.3 (HM)) | 31 | ||||||
| XZ-301265-2 (M) | c.2291G>A | ND | 6 | c.2291G>A | 2.6 (LP) | 2.6 (LP) | 96 | EORD + nystagmus | No | No | No | No |
| p.Arg764His | 34 | 6 | p.Arg764His | 2.6 (LP) | 2.6 (LP) | 42 | ||||||
| XZ-010621 | c.2234C>A | 0.5 | 8.75 | c.2843G>A | 0.7 (20/100) | 0.9 (20/160) | 228 | EORD + nystagmus | No | No | No | Yes |
| (M) | p.Thr745Lys | 12 | 8.75 | p.Cys948Tyr | 0.5 (20/63) | 0.6 (20/80) | 31 | |||||
| XZ-344444 | c.2498G>A | 0.5 | 9.5 | c.506del | 2.6 (LP) | 2.6 (LP) | 24 | EORD + nystagmus | Yes | No | No | No |
| (F) | p.Gly833Asp | 28 | 8.5 | p.Gly169Valfs*3 | 2.6 (LP) | 2.6 (LP) | 30 | |||||
| M-1861 | c.2639A>G | 5 | 0.25 | c.54_55insT | 1.5 (20/600) | ND | ND | EORD | Yes | Yes | No | No |
| (M) | p.Asn880Ser | 17 | 0 | p.Leu19Serfs*11 | 1.2 (20/320) | |||||||
| M-1732 | c.2401A>T | 3 | 9.25 | c.974G>T | 0.7 (20/100) | 0.7 (20/100) | 39 | EORD | Yes | No | No | No |
| (F) | p.Lys801* | 23.5 | 8.5 | p.Cys325Phe | 0.7 (20/100) | 0.7 (20/100) | 26.5 | |||||
| M-3530 | c.2234C>T | 3 | 2 | c.2234C>T | 1.0 (20/200) | 1.0 (20/200) | 36 | RP | Yes | Yes | No | No |
| (M) | p.Thr745Met | 25 | 2.5 | p.Thr745Met | 1.3 (20/400) | 1.0 (20/200) | 28 | |||||
| XZ-381491 | c.2843G>A | 9 | 7.75 | c.3307G>A | 0.6 (20/80) | 2.6 (LP) | 12 | RP | No | No | No | No |
| (F) | p.Cys948Tyr | 26 | 7 | p.Gly1103Arg | 0.9 (20/160) | 2.6 (LP) | 27 | |||||
| M-731 | c.2290C>T | 6 | 4 | c.3308G>T | 0.5 (20/63) | 0.8 (20/125) | 182 | RP | No | Yes | No | Yes |
| (M) | p.Arg764Cys | 8 | 4.25 | p.Gly1103Val | 0.4 (20/50) | 0.7 (20/100) | 23 | |||||
| M-2804-1 | c.2308G>A | 9 | 1.5 | c.2308G>A | 0.4 (20/50) | 0.4 (20/50) | 32 | RP | No | No | No | Yes |
| (M) | p.Gly770Ser | 9.5 | 1 | p.Gly770Ser | 0.3 (20/40) | 0.4 (20/50) | 12 | |||||
| M-2804-2 | c.2308G>A | 7 | ND | c.2308G>A | ND | ND | ND | RP | No | No | No | Yes |
| (F) | p.Gly770Ser | 32 | p.Gly770Ser | |||||||||
| M-2121 | c.1817G>C | 14 | 5.5 | c.613_619del | 0.4 (20/50) | 0.4 (20/50) | 37 | RP | No | No | No | Yes |
| (M) | p.Cys606Ser | 17.5 | 5.5 | p.Ile205Aspfs*13 | 0.4 (20/50) | 0.4 (20/50) | 21 | |||||
| M-2415 | c.2843G>A | 6 | −0.25 | c.2423A>G | 0.2 (20/32) | 0.7 (20/100) | 54 | RP | Yes | No | No | Yes |
| (F) | p.Cys948Tyr | 23.5 | −0.25 | p.Tyr808Cys | 0.2 (20/32) | 0.4 (20/50) | 28 | |||||
| M-699 | c.2290C>T | 7 | ND | c.2290C>T | 2.6 (LP) | ND | ND | RP | ND | ND | ND | ND |
| (F) | p.Arg764Cys | 80 | p.Arg764Cys | 2.6 (LP) | ||||||||
| M-4075 | c.3299>C | 6 | 0.75 | c.653-1G>T | 0.4 (20/50) | ND | ND | RP | No | No | No | Yes |
| (F) | p.Ile1100Thr | 7 | 0.75 | p.? | 0.1 (20/25) | |||||||
| M-4463 | c.2843G> A | 6 | ND | c.2843G> A | 2.6 (LP) | ND | ND | RP | No | No | No | No |
| (F) | p. Cys948Tyr | 48 | ND | p.Cys948Tyr | 2.6 (LP) | |||||||
| M-4570-1 | c.2506C>A | 6 | 3 | c.2290C>T | 0.2 (20/32) | 0.2 (20/32) | 29 | RP | No | No | No | Yes |
| (F) | p.Pro836Thr | 10 | 3 | p.Arg764Cys | 0.5 (20/63) | 0.7 (20/100) | 12 | |||||
| M–3915 | c.613_619del | >5 | ND | c.3482A>G | 0.7 (20/100) | ND | ND | RP | No | No | No | Yes |
| (M) | p.Ile205Aspfs*13 | 61.5 | −0.25 | p.Tyr1161Cys | 0.2 (20/32) | |||||||
| XZ-332989 | c.2549G>C | 5 | 5 | c.2585T>G p.Leu862Arg | 0.6 (20/80) | 0.8 (20/125) | 108 | RP | No | No | No | Yes |
| (F) | p.Gly850Arg | 7 | 5.25 | 0.6 (20/80) | 0.8 (20/125) | 16 | ||||||
| L-93112991 (M) | c.2843G>A | ND | −1.75 | c.498_506del | 0.5 (20/63) | ND | ND | MD | No | No | No | ND |
| p.Cys948Tyr | 44 | −1.50 | p.Ile167_Gly169del | 0.7 (20/100) | ||||||||
| L-13010924 (M) | c.2843G>A | 22 | 0.75 | c.498_506del | 0.6 (20/80) | 1.5 (20/600) | 60 | MD | No | No | No | No |
| p.Cys948Tyr | 23 | 0.25 | p.Ile167_Gly169del | 0.2 (20/32) | 0.4 (20/50) | 28 | ||||||
| M-1640-4 | c.1562C>T | 42 | −1.00 | c.1562C>T | 0.7 (20/100) | 0.5 (20/63) | 36 | MD | No | No | No | No |
| (F) | p.Ala521Val | 42 | −1.25 | p.Ala521Val | 1.0 (20/200) | 0.2 (20/32) | 45 | |||||
| M-3989 | c.2843G>A | 17 | −0.50 | c.498_506del | 1.3 (20/400) | ND | ND | MD | No | No | No | Yes |
| (M) | p.Cys948Tyr | 33 | −1.50 | p.Ile167_Gly169del | 0.1 (20/25) | |||||||
| M-2183 | c.946T>C | 21 | 3.25 | c.498_506del | 2.3 (HM) | ND | ND | MD | No | No | No | No |
| (M) | p.Cys316Arg | 33 | 3.25 | p.Ile167_Gly169del | 2.3 (HM) | |||||||
| M-3073-1 | c.2401A>T | 34 | −1.00 | c.498_506del | 0.5 (20/63) | ND | ND | MD | No | No | No | No |
| (M) | p.Lys801* | 37 | −0.50 | p.Ile167_Gly169del | 0.4 (20/50) | |||||||
| M-3073-3 | c.2401A>T | ND | ND | c.498_506del | ND | ND | ND | MD | ND | ND | ND | ND |
| (F) | p.Lys801* | 46 | p.Ile167_Gly169del | |||||||||
| M-3377 | c.3014A>T | 40 | −0.50 | c.498_506del | 1.3 (20/400) | ND | ND | MD | No | No | No | Yes |
| (M) | p.Asp1005Val | 53.5 | −0.75 | p.Ile167_Gly169del | 1.3 (20/400) | |||||||
| M-3343 | c.498_506del | 16 | 0.5 | c.4142C>G | 0.4 (20/50) | ND | ND | MD | No | No | No | No |
| (M) | p.Ile167_Gly169del | 29.5 | 0.5 | p.Pro1381Arg | 0.7 (20/100) |
| N Eyes | Fovea Mean ± (SD) | T1500 Mean ± (SD) | T3000 Mean ± (SD) | N1500 Mean ± (SD) | ||
|---|---|---|---|---|---|---|
| Controls | Global | 28 | 226.79 (±20.71) | 321.46 (±19.03) | 257.89 (±17.08) | 347.21 (±19.29) |
| [0;10] years | 0 | - | - | - | - | |
| ]10;20] years | 2 | 266.50 (±0.71) | 310.00 (±2.83) | 260.00 (±1.41) | 341.00 (±1.41) | |
| ]20;30] years | 10 | 218.30 (±14.58) | 322.90 (±18.04) | 261.40 (±12.31) | 352.50 (±21.08) | |
| ]30;40] years | 4 | 234.25 (±20.65) | 305.75 (±6.55) | 242.50 (±2.38) | 330.25 (±13.74) | |
| ]40;50] years | 4 | 204.25 (±7.32) | 312.25 (±6.70) | 244.25 (±7.37) | 337.50 (±9.54) | |
| >50 years | 8 | 235.00 (±14.92) | 335.00 (±22.37) | 267.50 (±23.19) | 355.50 (±19.89) | |
| Maculopathy | Global | 14 | 106.57 (±23.64) | 284.43 (±47.00) | 232.36 (±28.32) | 308.79 (±33.24) |
| [0;10] years | 0 | - | - | - | - | |
| ]10;20] years | 0 | - | - | - | - | |
| ]20;30] years | 4 | 108.00 (±5.29) | 248.50 (±31.27) | 214.00 (±28.01) | 281.00 (±14.88) | |
| ]30;40] years | 6 | 96.33 (±21.92) | 299.67 (±45.45) | 255.00 (±20.28) | 335.00 (±23.16) | |
| ]40;50] years | 2 | 108.00 (±1.41) | 263.00 (±9.90) | 208.50 (±2.12) | 304.00 (±36.77) | |
| >50 years | 2 | 133.00 (±52.33) | 332.00 (±57.98) | 225.00 (±18.38) | 290.50 (±41.72) | |
| Retinitis Pigmentosa | Global | 21 | 272.95 (±150.51) | 353.48 (±49.72) | 344.76 (±75.15) | 382.67 (±42.48) |
| [0;10] years | 8 | 296.50 (±211.71) | 360.88 (±67.27) | 358.38 (±100.29) | 396.88 (±53.90) | |
| ]10;20] years | 4 | 266.75 (±112.23) | 356.75 (±12.34) | 343.00 (±38.76) | 372.00 (±21.02) | |
| ]20;30] years | 6 | 242.67 (±115.77) | 362.50 (±35.14) | 364.83 (±58.33) | 384.17 (±38.92) | |
| ]30;40] years | 0 | - | - | - | - | |
| ]40;50] years | 1 | 177.00 * | 257.00 * | 250.00 * | 314.00 * | |
| >50 years | 2 | 330.00 (±93.34) | 338.50 (±20.51) | 281.00 (±4.24) | 377.00 (±0.00) | |
| EORD | Global | 17 | 268.94 (±169.17) | 363.71 (±55.83) | 332.53 (±58.25) | 415.29 (±70.12) |
| [0;10] years | 10 | 290.30 (±201.05) | 388.10 (±51.25) | 355.10 (±61.94) | 434.00 (±71.05) | |
| ]10;20] years | 2 | 403.00 (±21.21) | 342.50 (±3.54) | 318.00 (±12.73) | 464.50 (±6.36) | |
| ]20;30] years | 2 | 190.50 (±61.52) | 374.00 (±18.38) | 324.50 (±30.41) | 404.50 (±27.58) | |
| ]30;40] years | 1 | 185.00 * | 311.00 * | 257.00 * | 348.00 * | |
| ]40;50] years | 0 | - | - | - | - | |
| >50 years | 2 | 148.50 (±31.82) | 279.00 (±41.01) | 280.00 (±42.43) | 317.00 (±1.41) |
| Patient ID | Allele 1 | Impact on CRB1-A | Impact on CRB1-B | Allele 2 | Impact on CRB1-A | Impact on CRB1-B | Müller Cell Isoform | Photoreceptor Isoform |
|---|---|---|---|---|---|---|---|---|
| M-1640-1 | p.Ala521Val | Laminin G-like 1 | Laminin G-like 1 | p.Ala521Val | Laminin G-like 1 | Laminin G-like 1 | 100% mutated | 100% mutated |
| M-2129 | p.Thr745Met | Laminin G-like 2 | Laminin G-like 2 | p.Thr745Met | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-567 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Ile482_Thr484del | Deletion between EGF-like 11 and Lam G-like 1 | Deletion between EGF-like 11 and Lam G-like 1 | 100% mutated | 100% mutated |
| M-116 | p.Trp229* | NMD | WT | p.Ter1407Lysext*111 | Cytoplasmic C-term | WT | 50% mutated/0% WT | 100% WT |
| M-3361 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-3591 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Lys801* | NMD | NMD | 50% mutated/0% WT | 50% mutated/0% WT |
| M-2897 | p.Gly850Ser | Laminin G-like 2 | Laminin G-like 2 | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 100% mutated |
| M-3235 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Leu1405Argfs*82 | Cytoplasmic C-term | WT | 100% mutated | 50% mutated/50% WT |
| M-1316-4 | p.Pro748Hisfs*6 | NMD | NMD | p.Pro748Hisfs*6 | NMD | NMD | No protein | No protein |
| M-340 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Gly827* | NMD | NMD | 50% mutated/0% WT | 50% mutated/0% WT |
| L- 15090708 | p.Lys801* | NMD | NMD | p.Cys1238Phe | EGF-like-17 | EGF-like-17 | 50% mutated/0% WT | 50% mutated/0% WT |
| L-14061710 | p.Gly850Ser | Laminin G-like 2 | Laminin G-like 2 | p.Pro1381Arg | Cytoplasmic C-term | WT | 100% mutated | 50% mutated/50% WT |
| XZ-372875 | p.Cys1229Ser | EGF-like 19 | EGF-like 19 | p.Leu1107Pro | Laminin G-like 3 | Laminin G-like 3 | 100% mutated | 100% mutated |
| M-1316-1 | p.Pro748Hisfs*6 | NMD | NMD | p.Pro748Hisfs*6 | NMD | NMD | No protein | No protein |
| M-3324 | p.Gln362* | NMD | WT | p.Lys801* | NMD | NMD | No protein | 0% mutated/50% WT |
| M-2427 | p.Thr745Met | Laminin G-like 2 | Laminin G-like 2 | p.Ile1358Asn | Cytoplasmic C-term | WT | 100% mutated | 50% mutated/50% WT |
| M-4621-1 | p.Pro1381Arg | Cytoplasmic C-term | WT | p.Pro1381Arg | Cytoplasmic C-term | WT | 100% mutated | 100% WT |
| M-1580 | p.Val1055Glu | Laminin G-like 3 | Laminin G-like 3 | p.Leu1107Pro | Laminin G-like 3 | Laminin G-like 3 | 100% mutated | 100% mutated |
| M-2123 | c.3999_4005 + 4delinsAAAGGAGAGC | ? | Cytoplasmic C-term | c.3999_4005 + 4delinsAAAGGAGAGC | ? | Cytoplasmic C-term | 100% mutated | 100% mutated |
| M-2123-2 | c.3999_4005 + 4delinsAAAGGAGAGC | ? | Cytoplasmic C-term | c.3999_4005 + 4delinsAAAGGAGAGC | ? | Cytoplasmic C-term | 100% mutated | 100% mutated |
| M-2186 | p.Asn657Lysfs*8 | NMD | NMD | p.Asn657Lysfs*8 | NMD | NMD | No protein | no protein |
| M-2184 | p.Lys801* | NMD | NMD | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 50% mutated/0% WT | 50% mutated/0% WT |
| XZ-301265-1 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| XZ-301265-2 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| XZ-010621 | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | p.Thr745Lys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| XZ-344444 | p.Gly169Valfs*3 | NMD | WT | p.Gly833Asp | Laminin G-like 2 | Laminin G-like 2 | 50% mutated/0% WT | 50% mutated/50% WT |
| M-1861 | p.Leu19Serfs*11 | NMD | WT | p.Asn880Ser | Laminin G-like 2 | Laminin G-like 2 | 50% mutated/0% WT | 50% mutated/50% WT |
| M-1732 | p.Cys325Phe | EGF-like 8 | WT | p.Lys801* | NMD | NMD | 50% mutated/0% WT | 0% mutated/50% WT |
| M-3530 | p.Thr745Met | Laminin G-like 2 | Laminin G-like 2 | p.Thr745Met | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| XZ-381491 | p.Gly1103Arg | Laminin G-like 3 | Laminin G-like 3 | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 100% mutated |
| M-731 | p.Gly1103Val | Laminin G-like 3 | Laminin G-like 3 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-2804-1 | p.Gly770Ser | Laminin G-like 2 | Laminin G-like 2 | p.Gly770Ser | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-2804-2 | p.Gly770Ser | Laminin G-like 2 | Laminin G-like 2 | p.Gly770Ser | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-2121 | p.Ile205Aspfs*13 | NMD | WT | p.Cys606Ser | Laminin G-like 1 | Laminin G-like 1 | 50% mutated/0% WT | 50% mutated/50% WT |
| M-2415 | p.Tyr808Cys | Laminin G-like 2 | Laminin G-like 2 | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 100% mutated |
| M-699 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-4075 | c.653-1G > T | ? | WT | p.Ile1100Thr | Laminin G-like 3 | Laminin G-like 3 | Unknown | 50% mutated/50% WT |
| M-4463 | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | p. Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 100% mutated |
| M-4570-1 | p.Arg764Cys | Laminin G-like 2 | Laminin G-like 2 | p.Pro836Thr | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| M-3915 | p.Tyr1161Cys | EGF-like 15 | EGF-like 15 | p.Ile205Aspfs*13 | NMD | WT | 50% mutated/0% WT | 50% mutated/50% WT |
| XZ-332989 | p.Leu862Arg | Laminin G-like 2 | Laminin G-like 2 | p.Gly850Arg | Laminin G-like 2 | Laminin G-like 2 | 100% mutated | 100% mutated |
| L-93112991 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 50% mutated/50% WT |
| L-13010924 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 50% mutated/50% WT |
| M-1640-4 | p.Ala521Val | Laminin G-like 1 | Laminin G-like 1 | p.Ala521Val | Laminin G-like 1 | Laminin G-like 1 | 100% mutated | 100% mutated |
| M-3989 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Cys948Tyr | EGF-like 14 | EGF-like 14 | 100% mutated | 50% mutated/50% WT |
| M-2183 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Cys316Arg | EGF-like 8 | WT | 100% mutated | 100% WT |
| M-3073-1 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Lys801* | NMD | NMD | 50% mutated/0% WT | 0% mutated/50% WT |
| M-3073-3 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Lys801* | NMD | NMD | 50% mutated/0% WT | 0% mutated/50% WT |
| M-3377 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Asp1005Val | Laminin G-like 3 | Laminin G-like 3 | 100% mutated | 50% mutated/50% WT |
| M-3343 | p.Ile167_Gly169del | EGF-like 4 | WT | p.Pro1381Arg | Cytoplasmic C-term | WT | 100% mutated | 100% WT |
| cDNA | Protein | Exon | Type of Variant | Gnomad | Softwares Prediction | ACMG Classification | Domain |
|---|---|---|---|---|---|---|---|
| c.54_55insT | p.Leu19Serfs*11 | 1 | Indel | Absent | Damaging | P | Signal peptide |
| c.687G>A | p.Trp229* | 3 | Nonsense | Absent | Damaging | P | EGF-like-6 |
| c.946T>C | p.Cys316Arg | 4 | Missense | Absent | Damaging for 23/1 | LP | EGF-like-8 |
| c.974G>T | p.Cys325Phe | 4 | Missense | Absent | Damaging for 22/2 | LP | EGF-like-8 |
| c.1445_1453del | p.Ile482_Thr484del | 6 | Del | Absent | Damaging | LP | ND |
| c.1562C>T | p.Ala521Val | 6 | Missense | Absent | Damaging for 8/15 | LP | Laminin G-like-1 |
| c.1817G>C | p.Cys606Ser | 6 | Missense | Absent | Damaging for 8/16 | LP in trans | Laminin G-like-1 |
| c.1971del | p.Asn657Lysfs*8 | 6 | Del | Absent | Damaging | P | Laminin G-like-1 |
| c.2243del | p.Pro748Hisfs*6 | 7 | Del | Absent | Damaging | P | Laminin G-like-2 |
| c.2423A>G | p.Tyr808Cys | 7 | Missense | Absent | Damaging for 6/18 | LP in trans | Laminin G-like-2 |
| c.2549G>C | p.Gly850Arg | 7 | Missense | Absent | Damaging for 21/3 | LP | Laminin G-like-2 |
| c.2585T>G | p.Leu862Arg | 7 | Missense | Absent | Damaging for 22/2 | LP | Laminin G-like-2 |
| c.3164T>A | p.Val1055Glu | 9 | Missense | Absent | Damaging for 18/6 | LP | Laminin G-like-3 |
| c.3713G>T | p.Cys1238Phe | 9 | Missense | Absent | Damaging for 23/1 | LP | EGF-like-17 |
| c.3999_4005 + 4delinsAAAGGAGAGC | p.? | 11 | Indel | Absent | Damaging | P | EGF-like-18/TM domain |
| c.4214del | p.Leu1405Argfs*82 | 12 | Del | Absent | Damaging | LP | Intracellular tail |
| c.4219T > A | p.Ter1407Lysext*111 | 12 | Stop loss | Absent | Damaging for 3/5 | LP in trans | Intracellular tail |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mairot, K.; Smirnov, V.; Bocquet, B.; Labesse, G.; Arndt, C.; Defoort-Dhellemmes, S.; Zanlonghi, X.; Hamroun, D.; Denis, D.; Picot, M.-C.; et al. CRB1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Müller Cell and Photoreceptor CRB1 Isoforms. Int. J. Mol. Sci. 2021, 22, 12642. https://doi.org/10.3390/ijms222312642
Mairot K, Smirnov V, Bocquet B, Labesse G, Arndt C, Defoort-Dhellemmes S, Zanlonghi X, Hamroun D, Denis D, Picot M-C, et al. CRB1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Müller Cell and Photoreceptor CRB1 Isoforms. International Journal of Molecular Sciences. 2021; 22(23):12642. https://doi.org/10.3390/ijms222312642
Chicago/Turabian StyleMairot, Kévin, Vasily Smirnov, Béatrice Bocquet, Gilles Labesse, Carl Arndt, Sabine Defoort-Dhellemmes, Xavier Zanlonghi, Dalil Hamroun, Danièle Denis, Marie-Christine Picot, and et al. 2021. "CRB1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Müller Cell and Photoreceptor CRB1 Isoforms" International Journal of Molecular Sciences 22, no. 23: 12642. https://doi.org/10.3390/ijms222312642
APA StyleMairot, K., Smirnov, V., Bocquet, B., Labesse, G., Arndt, C., Defoort-Dhellemmes, S., Zanlonghi, X., Hamroun, D., Denis, D., Picot, M.-C., David, T., Grunewald, O., Pégart, M., Huguet, H., Roux, A.-F., Kalatzis, V., Dhaenens, C.-M., & Meunier, I. (2021). CRB1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Müller Cell and Photoreceptor CRB1 Isoforms. International Journal of Molecular Sciences, 22(23), 12642. https://doi.org/10.3390/ijms222312642