
Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
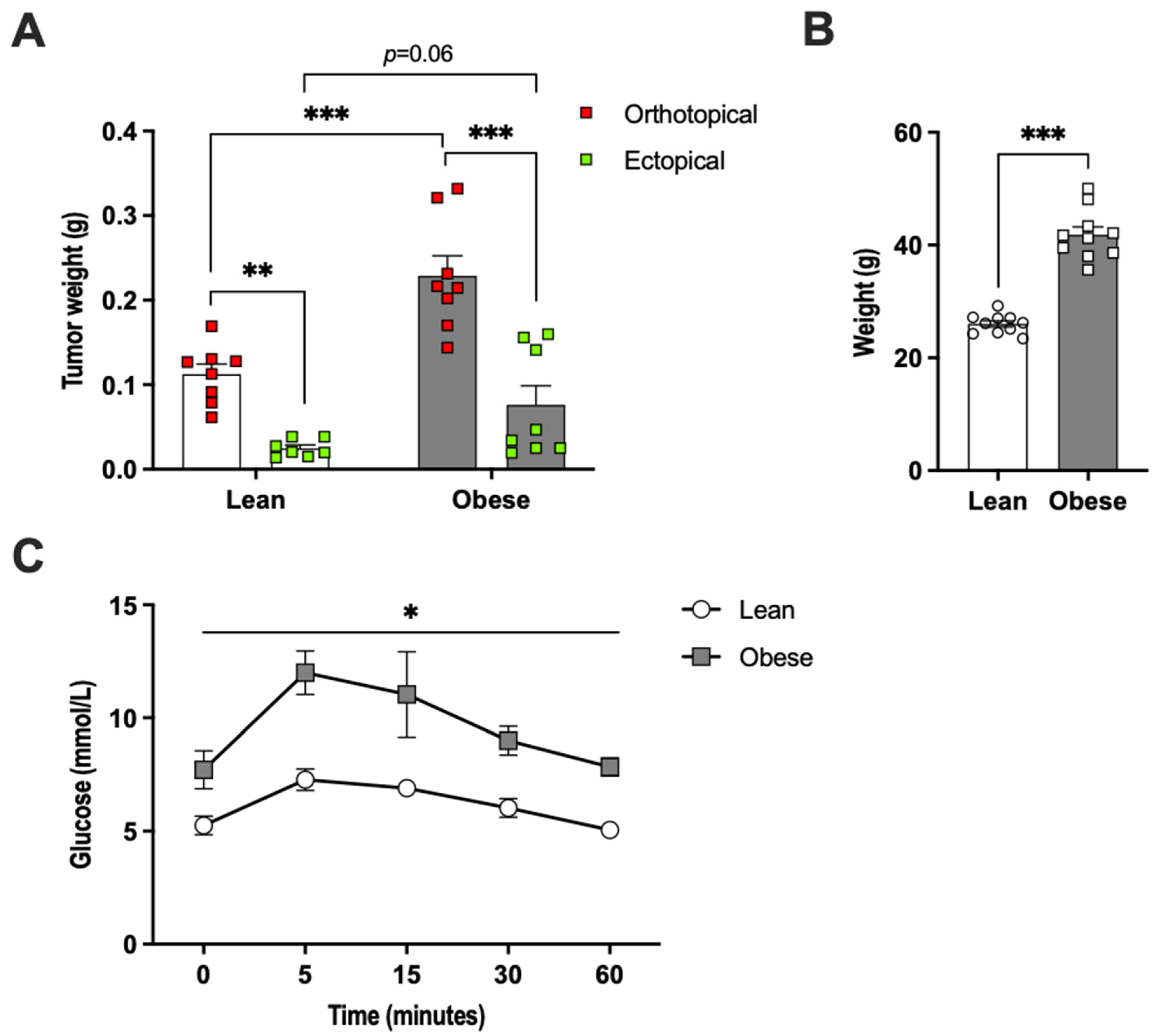
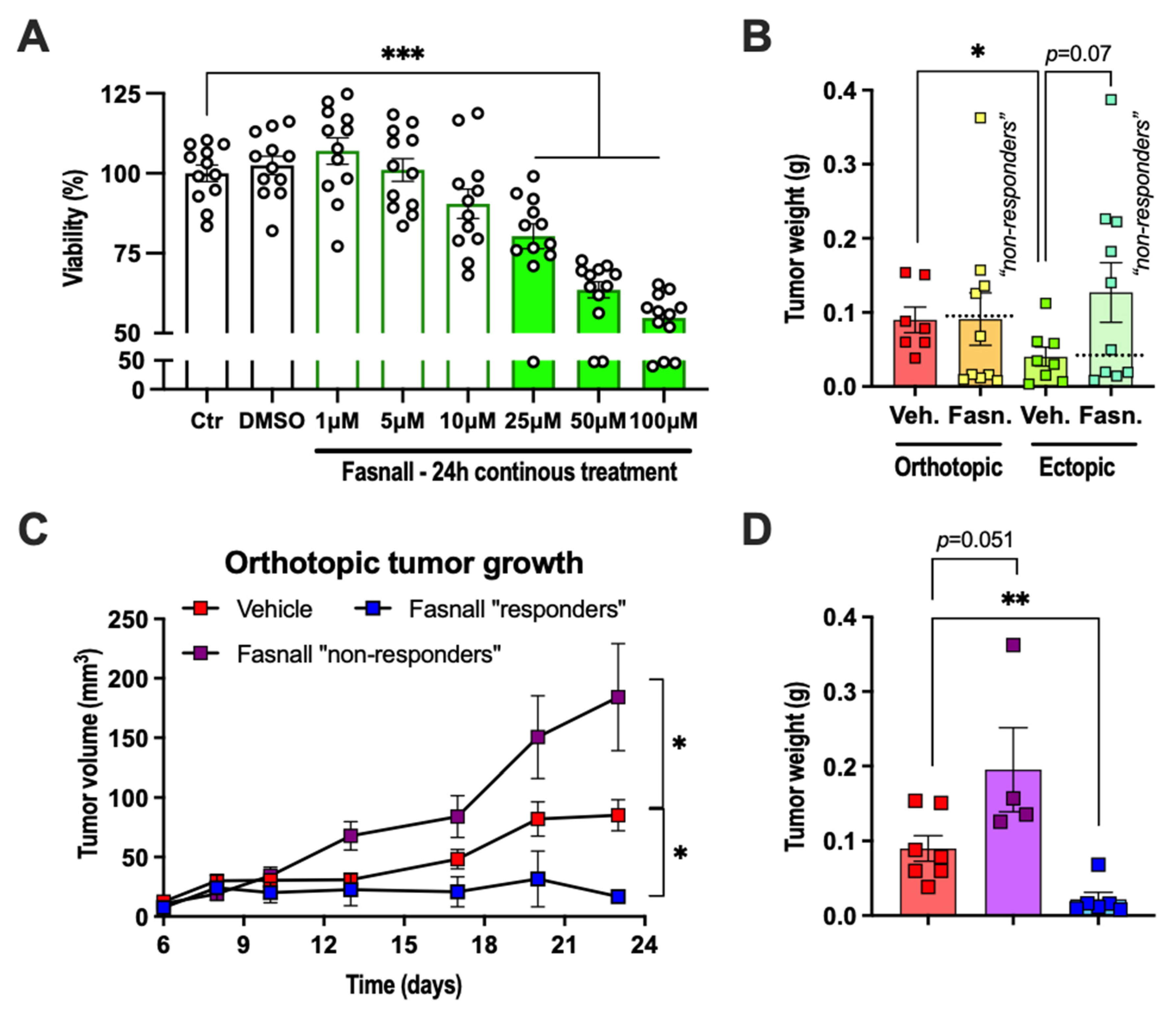
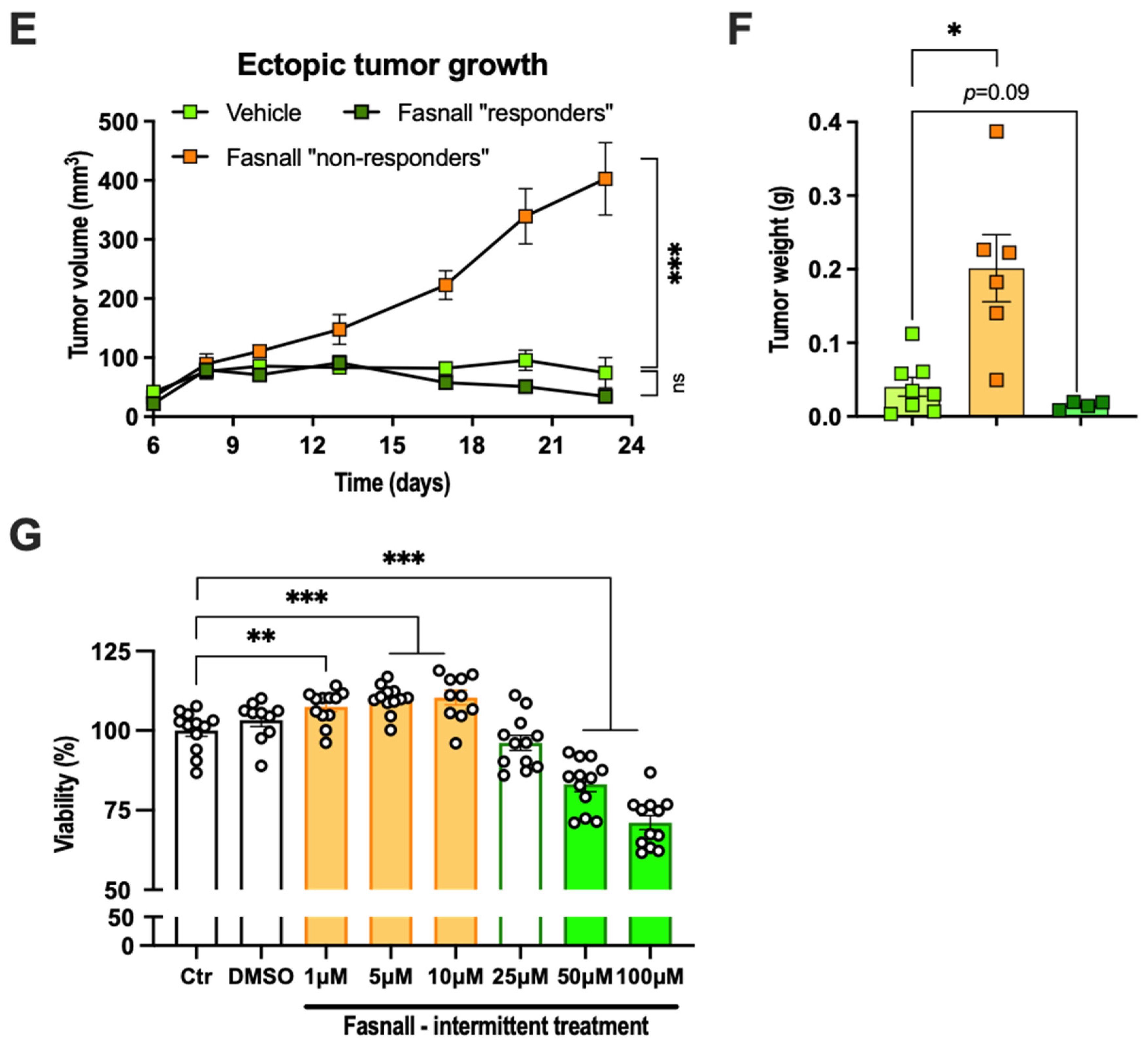
2.1. Adipose Tissue Accelerated E0771 Breast Cancer Growth In Vivo
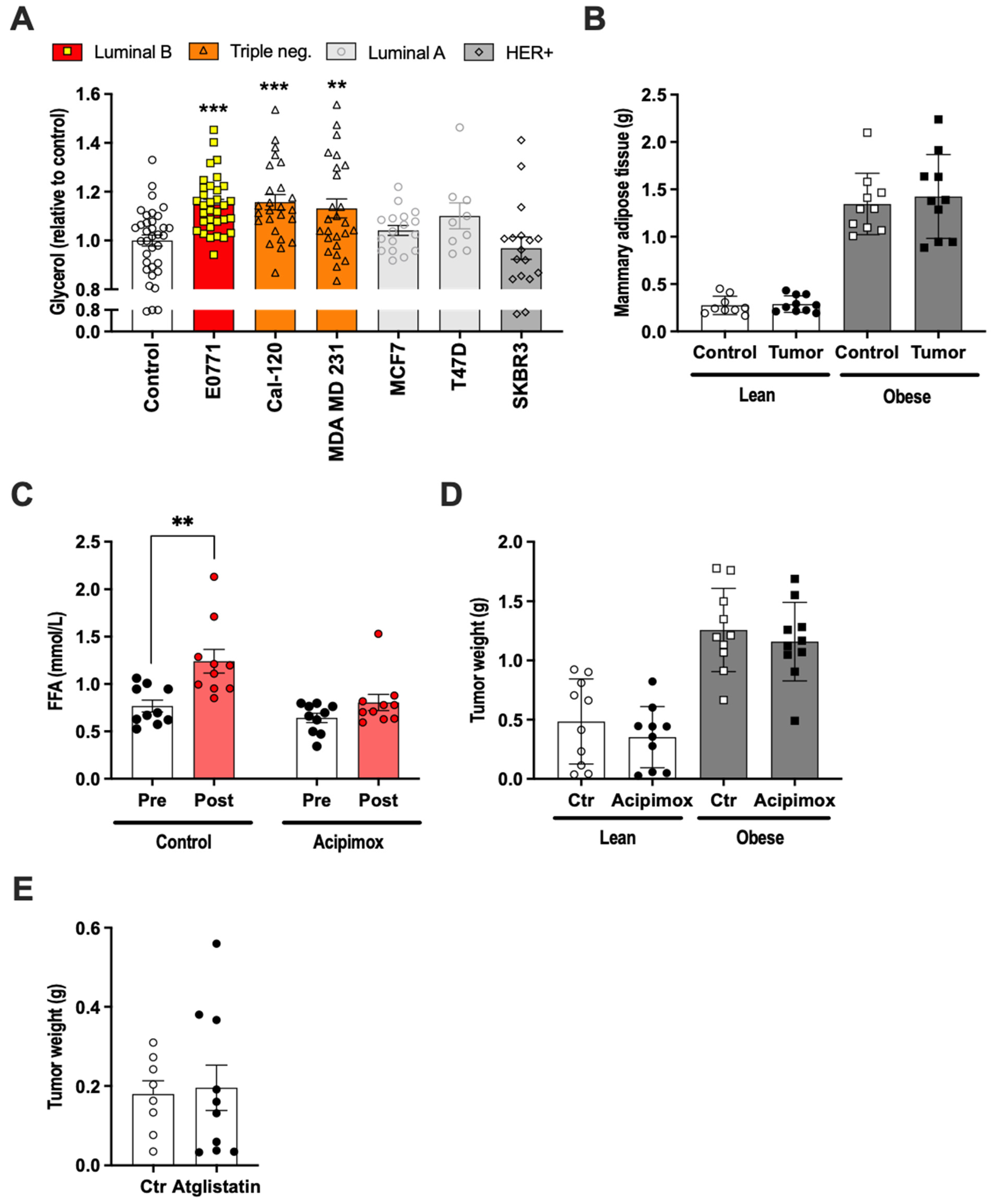
2.2. E0771 Breast Cancer Induced Increased Lipolysis, but Lipolysis Inhibitor Treatment Had No Effect on E0771 Tumor Growth In Vivo
2.3. The E0771 Tumor Growth-Promoting Effect of Adipose Tissue Was Associated with Increased Tumor Lipogenesis
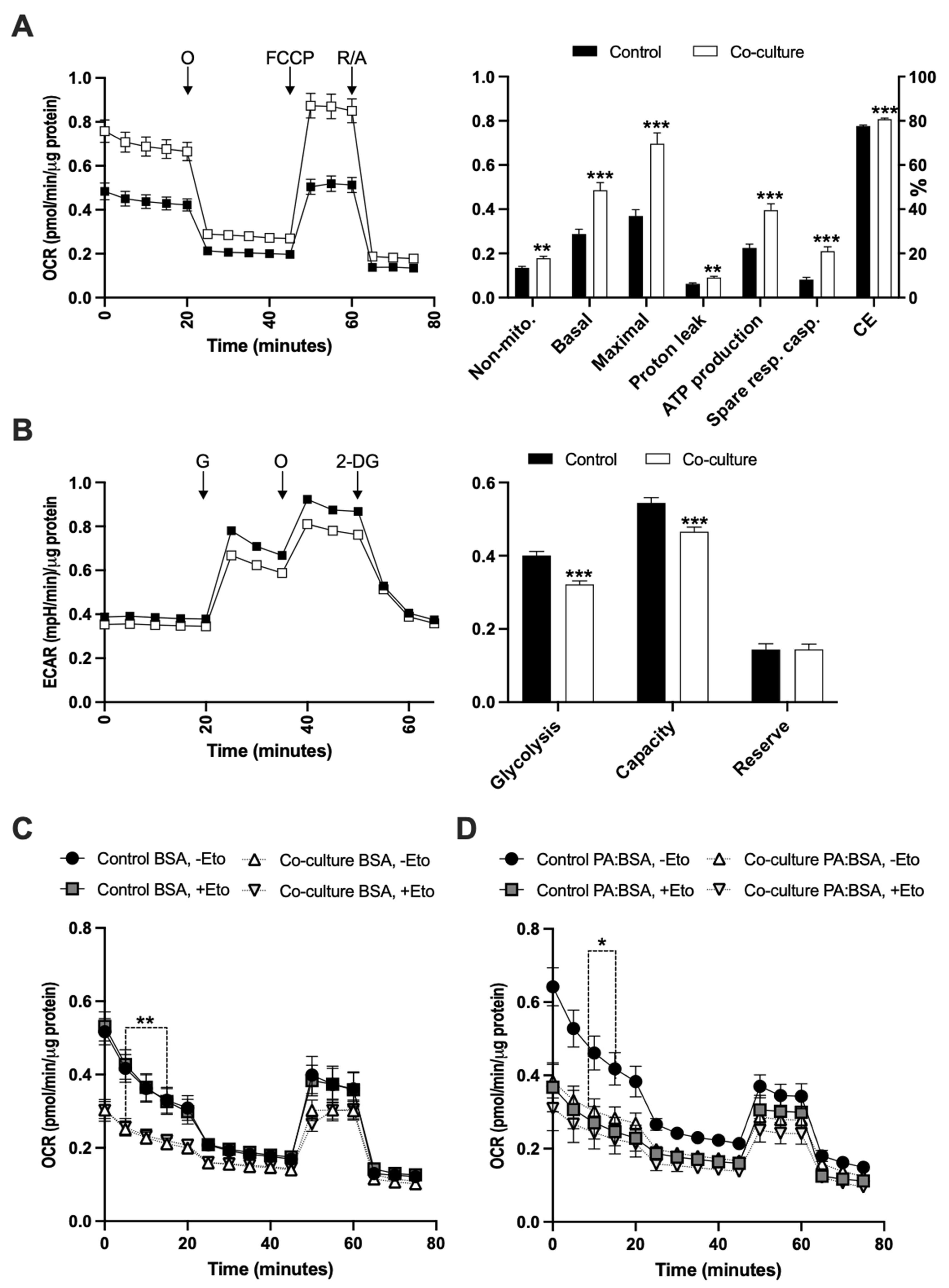
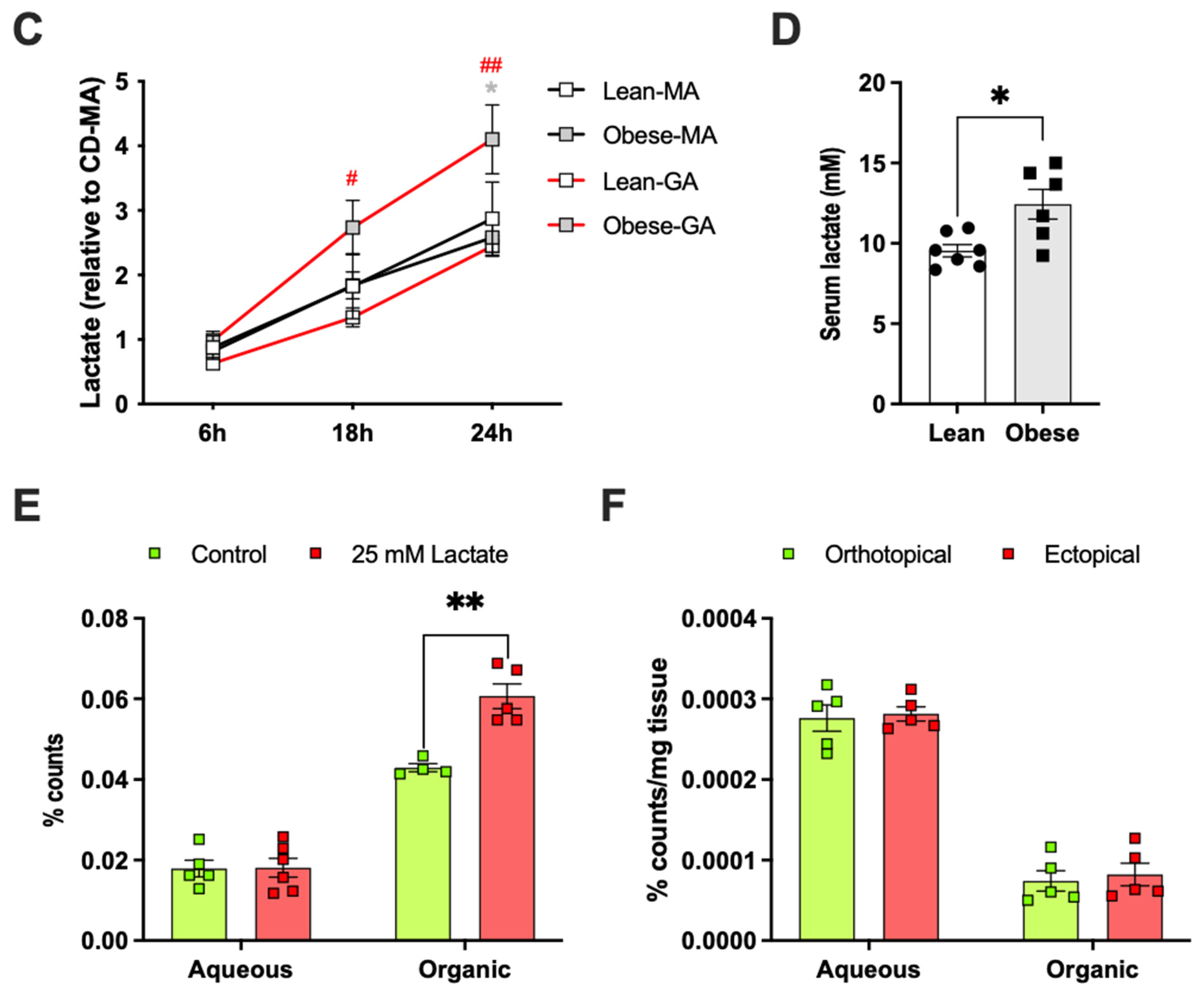
2.4. Adipose Tissue Co-Culture Led to Increased Oxygen Consumption and Reduced Glycolysis in E0771 Cells
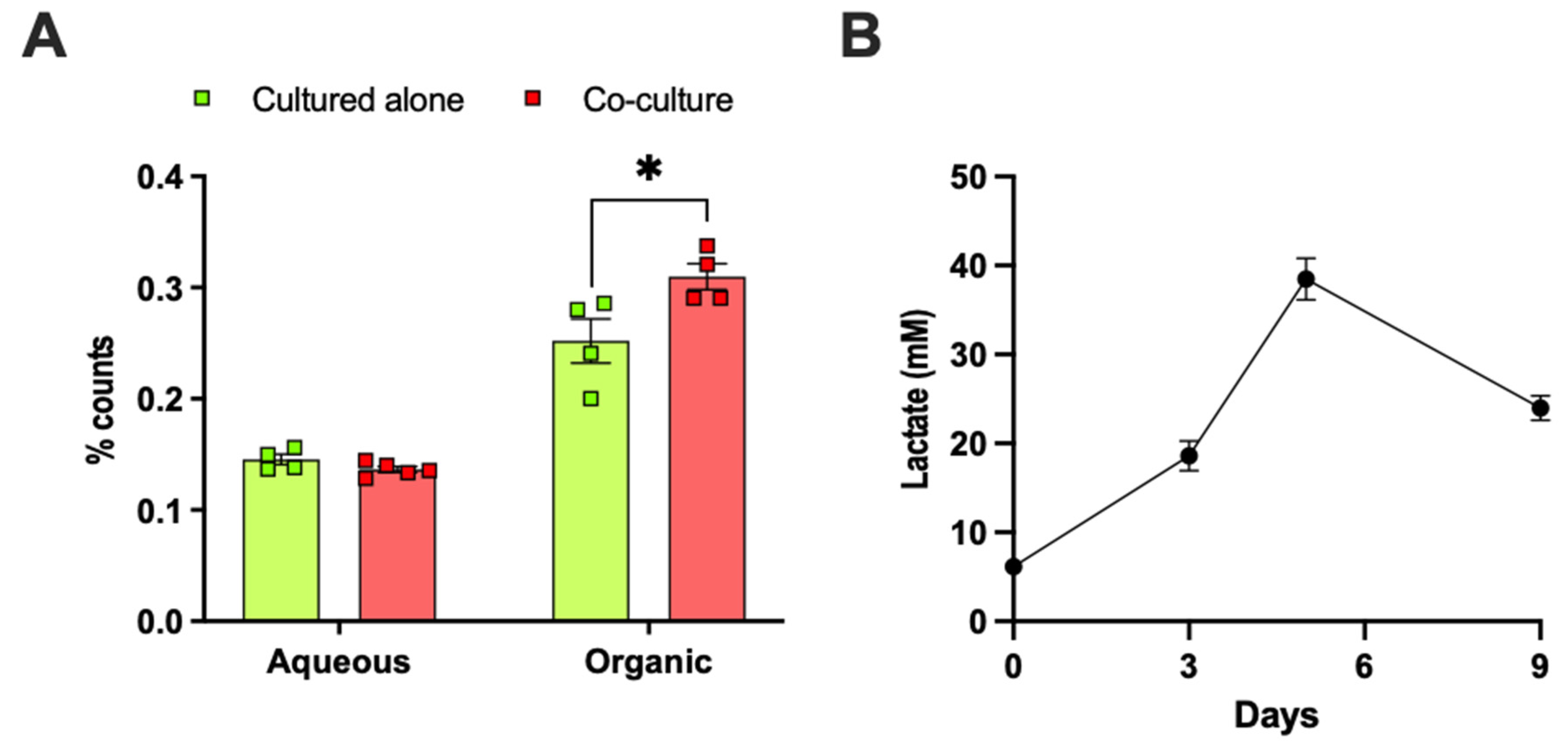
2.5. Adipose Tissue Co-Culture or Lactate Pretreatment Led to Increased De Novo Lipogenesis from Glucose in E0771 Cells
2.6. Dichotomous Effect of Pharmacological FASN Inhibition on E0771 Growth In Vivo and Viability In Vitro
3. Discussion
3.1. Lipid Metabolism in Orthotopic E0771 Tumors—Metabolic Symbiosis with Adipose Tissue
3.2. Lipolysis and Lipogenesis as Therapeutic Targets in Breast Cancer Treatment
4. Materials and Methods
4.1. Animals
4.2. E0771 Breast Cancer Model
4.3. Lipolysis Inhibitor Treatment In Vivo
4.4. Cell Lines and Cell Culture
4.5. Conditioned Medium Preparation
4.6. Lipolysis In Vitro
4.7. Lipid, Glucose and Lactate Metabolism In Vivo
4.8. RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR
4.9. E0771 and Adipose Tissue Co-Culture Studies
4.10. Characterization of Mitochondrial Function and Glycolysis
4.11. De Novo Lipogenesis In Vitro
4.12. Lipogenesis Inhibitor Treatment
4.13. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. Prog. Mol. Biol. Transl. 2017, 151, 1–32. [Google Scholar]
- Fallahpour, S.; Navaneelan, T.; De, P.; Borgo, A. Breast cancer survival by molecular subtype: A population-based analysis of cancer registry data. CMAJ Open 2017, 5, E734–E739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaaskelainen, A.; Roininen, N.; Karihtala, P.; Jukkola, A. High Parity Predicts Poor Outcomes in Patients With Luminal B-Like (HER2 Negative) Early Breast Cancer: A Prospective Finnish Single-Center Study. Front. Oncol. 2020, 10, 1470. [Google Scholar] [CrossRef]
- Ades, F.; Zardavas, D.; Bozovic-Spasojevic, I.; Pugliano, L.; Fumagalli, D.; de Azambuja, E.; Viale, G.; Sotiriou, C.; Piccart, M. Luminal B breast cancer: Molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 2794–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.C.; Church, F.C. Mature breast adipocytes promote breast cancer cell motility. Exp. Mol. Pathol. 2012, 92, 312–317. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Buache, E.; Chenard, M.P.; Dali-Youcef, N.; Rio, M.C. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int. J. Dev. Biol. 2011, 55, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Lehuede, C.; Laurent, V.; Dirat, B.; Dauvillier, S.; Bochet, L.; Le Gonidec, S.; Escourrou, G.; Valet, P.; Muller, C. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012, 324, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.C.; Hsiao, M. Leptin and Cancer: Updated Functional Roles in Carcinogenesis, Therapeutic Niches, and Developments. Int. J. Mol. Sci. 2021, 22, 2870. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Al-Bahlani, S.; Al-Lawati, H.; Al-Adawi, M.; Al-Abri, N.; Al-Dhahli, B.; Al-Adawi, K. Fatty acid synthase regulates the chemosensitivity of breast cancer cells to cisplatin-induced apoptosis. Apoptosis 2017, 22, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Ford, J.H. Saturated fatty acid metabolism is key link between cell division, cancer, and senescence in cellular and whole organism aging. Age 2010, 32, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R.; Colomer, R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER-2/neu-overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res. Treat. 2004, 84, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Ropero, S.; Brunet, J.; Colomer, R.; Menendez, J.A. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol. Rep. 2007, 18, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Mehmi, I.; Papadimitropoulou, A.; Vander Steen, T.; Cuyas, E.; Verdura, S.; Espinoza, I.; Vellon, L.; Atlas, E.; Lupu, R. Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7661. [Google Scholar] [CrossRef] [PubMed]
- Sounni, N.E.; Cimino, J.; Blacher, S.; Primac, I.; Truong, A.; Mazzucchelli, G.; Paye, A.; Calligaris, D.; Debois, D.; De Tullio, P.; et al. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014, 20, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.M.; Colombo, I.; Montorfano, G.; Zava, S.; Corsetto, P.A. Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 2021, 10, 175. [Google Scholar] [CrossRef]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Naour, A.; Koffi, Y.; Diab, M.; Le Guennec, D.; Rouge, S.; Aldekwer, S.; Goncalves-Mendes, N.; Talvas, J.; Farges, M.C.; Caldefie-Chezet, F.; et al. EO771, the first luminal B mammary cancer cell line from C57BL/6 mice. Cancer Cell Int. 2020, 20, 328. [Google Scholar] [CrossRef]
- Casey, A.E.; Laster, W.R., Jr.; Ross, G.L. Sustained enhanced growth of carcinoma EO771 in C57 black mice. Proc. Soc. Exp. Biol. Med. 1951, 77, 358–362. [Google Scholar] [CrossRef]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Di Girolamo, M.; Newby, F.D.; Lovejoy, J. Lactate production in adipose tissue: A regulated function with extra-adipose implications. FASEB J. 1992, 6, 2405–2412. [Google Scholar] [CrossRef]
- Jansson, P.A.; Smith, U.; Lonnroth, P. Evidence for lactate production by human adipose tissue in vivo. Diabetologia 1990, 33, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Thacker, S.V.; Nickel, M.; DiGirolamo, M. Effects of food restriction on lactate production from glucose by rat adipocytes. Am. J. Physiol. 1987, 253, E336–E342. [Google Scholar] [CrossRef] [PubMed]
- Sabater, D.; Arriaran, S.; Romero Mdel, M.; Agnelli, S.; Remesar, X.; Fernandez-Lopez, J.A.; Alemany, M. Cultured 3T3L1 adipocytes dispose of excess medium glucose as lactate under abundant oxygen availability. Sci. Rep. 2014, 4, 3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krycer, J.R.; Quek, L.E.; Francis, D.; Fazakerley, D.J.; Elkington, S.D.; Diaz-Vegas, A.; Cooke, K.C.; Weiss, F.C.; Duan, X.; Kurdyukov, S.; et al. Lactate production is a prioritized feature of adipocyte metabolism. J. Biol. Chem. 2020, 295, 83–98. [Google Scholar] [CrossRef]
- Kalezic, A.; Udicki, M.; Srdic Galic, B.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Lactate Metabolism in Breast Cancer Microenvironment: Contribution Focused on Associated Adipose Tissue and Obesity. Int. J. Mol. Sci. 2020, 21, 9676. [Google Scholar] [CrossRef]
- Cheung, S.M.; Husain, E.; Masannat, Y.; Miller, I.D.; Wahle, K.; Heys, S.D.; He, J. Lactate concentration in breast cancer using advanced magnetic resonance spectroscopy. Br. J. Cancer 2020, 123, 261–267. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, M.J.; Li, Q.; Lee, S.; Zhang, C.; Kull, B.; Hallen, S.; Thorell, A.; Alexandersson, I.; Hagberg, C.E.; Peng, X.R.; et al. Mature Human White Adipocytes Cultured under Membranes Maintain Identity, Function, and Can Transdifferentiate into Brown-like Adipocytes. Cell Rep. 2019, 27, 213–225.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001, 2, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Tai, Y.; Zhou, J.; Gu, W.; Bai, Z.; Zhou, T.; Zhong, Z.; McCue, P.A.; Sang, N.; Ji, J.-Y. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle 2012, 11, 2348–2358. [Google Scholar] [CrossRef] [Green Version]
- Costa, V.; Foti, D.; Paonessa, F.; Chiefari, E.; Palaia, L.; Brunetti, G.; Gulletta, E.; Fusco, A.; Brunetti, A. The insulin receptor: A new anticancer target for peroxisome proliferator-activated receptor-g (PPARg) and thiazolidinedione-PPARg agonists. Endocr. Relat. Cancer 2008, 15, 325. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Millan, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer-Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2019, 9, 1536. [Google Scholar] [CrossRef]
- Christie, A.; McCormick, D.; Emmison, N.; Kraemer, F.; Alberti, K.; Yeaman, S. Mechanism of anti-lipolytic action of acipimox in isolated rat adipocytes. Diabetologia 1996, 39, 45–53. [Google Scholar]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785. [Google Scholar] [CrossRef] [Green Version]
- Green, H.; Kehinde, O. Sublines of Mouse 3t3 Cells That Accumulate Lipid. Cell 1974, 1, 113–116. [Google Scholar] [CrossRef]
- Kohn, A.D.; Summers, S.A.; Birnbaum, M.J.; Roth, R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996, 271, 31372–31378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carswell, K.A.; Lee, M.-J.; Fried, S.K. Culture of isolated human adipocytes and isolated adipose tissue. In Human Cell Culture Protocols; Springer: Berlin, Germany, 2012; pp. 203–214. [Google Scholar]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087379. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micallef, P.; Wu, Y.; Bauzá-Thorbrügge, M.; Chanclón, B.; Vujičić, M.; Peris, E.; Ek, C.J.; Wernstedt Asterholm, I. Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth. Int. J. Mol. Sci. 2021, 22, 11881. https://doi.org/10.3390/ijms222111881
Micallef P, Wu Y, Bauzá-Thorbrügge M, Chanclón B, Vujičić M, Peris E, Ek CJ, Wernstedt Asterholm I. Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth. International Journal of Molecular Sciences. 2021; 22(21):11881. https://doi.org/10.3390/ijms222111881
Chicago/Turabian StyleMicallef, Peter, Yanling Wu, Marco Bauzá-Thorbrügge, Belén Chanclón, Milica Vujičić, Eduard Peris, C. Joakim Ek, and Ingrid Wernstedt Asterholm. 2021. "Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth" International Journal of Molecular Sciences 22, no. 21: 11881. https://doi.org/10.3390/ijms222111881
APA StyleMicallef, P., Wu, Y., Bauzá-Thorbrügge, M., Chanclón, B., Vujičić, M., Peris, E., Ek, C. J., & Wernstedt Asterholm, I. (2021). Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth. International Journal of Molecular Sciences, 22(21), 11881. https://doi.org/10.3390/ijms222111881